Abstract
Hepatitis B x (HBx) is the smallest protein of 17kDa that is encoded by hepatitis B virus (HBV). HBx protein is more abundantly expressed in the cytoplasm than in the nucleus of HBV-infected hepatocytes. Considerable data suggest that HBx protein exploits the entire body of cellular signalling pathways by exerting its transcriptional transactivation activities and epigenetic alterations for viral survival and propagation. In this review, we intend to highlight the transcriptional activities of HBx protein and its role in inducing epigenetic anomalies that may lead to tumour initiation, aggressiveness and metastases.
Keywords
Epigenetics alterations; HBV; HBx; HCC; DNA Methylation; Histone Modification; Transcriptional activities
Abbreviations
CCCDNA: Covalently Closed Circular DNA; CUL4: Culin 4; COX-2: Cyclooxygenase 2; DDB1: Damage-Specific DNA Binding Protein 1; DNA: Deoxyribonucleic Acid; DLEC1: Deleted in Lung and Esophageal Cancer 1; DNMTs: DNA Methyltransferases; FLIP: FLICE Like Inhibitory Protein; HAT: Histone Acetyltransferase; HBV:
Hepatitis B Virus; HBx: Hepatitis B x; HBXIP: Hepatitis B-X Interacting Protein; HCC: Hepatocellular Carcinoma; HDACs: Histone Deacetylases; HIF-1 α: Hypoxia-Inducible Factor-1 Alpha; IL-8: Interleukin-8; JNK: Jun AminoTerminal Kinases; KDM1A: Lysine Specific Histone Demethylase 1; MEKK2: Mitogen-Activated Protein Kinase Kinase 2; miR: miRNA; NF-κB: Nuclear Factor Kappa B; PI3K: Phosphatidylinositol 3-Kinase; PTEN: Phosphatase and Tensin Homolog; SET1A: Su (var)3-9, Enhancer of zeste, Trithorax 1A; SFRP: Secreted Frizzled-Related Protein; STAT3: Signal Transducer and Activator of Transcription 3; SPHK1: Sphingosine Kinase 1; TGF-β1: Transforming Growth Factor Beta 1; TFAP2A: Transcription Factor AP-2 Alpha; VEGF: Vascular Endothelial Growth Factor.
Introduction
HBV is a non-cytopathic hepadnavirus transmitted prenatally, sexually and percutaneously. More than 350 million people are chronically infected worldwide and 1million die from infection annually as a result of hepatic cirrhosis and hepatocellular carcinoma (HCC) [1,2]. Amongst four HBV partially overlapping open reading frames is the smallest hepatitis B virus X (HBx) gene that encodes 154 amino acid regulatory protein [3]. HBx protein is a transcriptional transactivator that is required to initiate and maintain virus replication [4]. It promotes viral propagation and ultimately HBV-related malignant transformation by abnormally regulating several cellular pathways, which are involved in DNA repair, cell growth, differentiation, adhesion, proliferation and apoptosis [5-9]. This review highlights various molecular mechanisms which HBx protein utilises in promoting HBV-induced HCC.
Transcriptional Transactivation Activities of HBx Protein and Hepatocarcinogenesis
Dysregulated apoptosis is a phenotypic feature of HBV-related hepatocarcinogenesis. HBV-induced DNA damage often triggers localised apoptotic-related signals resulting in tissue necrosis. This process enables the elimination of damaged, unwanted and redundant hepatocytes that may otherwise lead to uncontrolled cell growth, proliferation and liver disease [10,11]. Aberrant regulation of cell proliferation and apoptosis has been identified as the consequence of abnormal inactivation or activation of gene transcription in HBV-induced HCC [10]. Although the underlying mechanism remains unknown, HBx protein may inhibit apoptosis by blocking the transactivation of caspase cascade 3, nuclear factor kappa B (NF-κB) and phosphatidylinositol 3-kinase (PI3K) signal transduction pathways [12-14]. For instance, HBx protein inhibits Fasmediated apoptosis of hepatoma cells by upregulating mFAS/FasL, sFas and NF-κB (Figure 1) [10,15]. On the other hand, interaction of HBV x-associated protein with protein kinase C was shown to induce apoptosis by switching on the transcription factor NF-κB [16,17]. Being localised in the cytoplasm, HBx protein also has the ability to sensitize HBV-infected cells towards FLICE-like inhibitory protein (FLIP), jun amino-terminal kinases (JNK), caspases 3 and 9 pro-apoptotic pathways and induce apoptosis [18]. HBx exerts its oncogenic properties and causes HCC in nude mice by transforming the non-transformed immortalized liver cell line QSG7701 [19]. HBx also communicates with mitogen activated protein kinases/extracellular signal regulated kinases (MAPK/ERK) signalling and activates PI3K/Akt pathway to transform cells leading to c-myc-mediated cell survival through inhibition of HBx-induced apoptosis (Figure 1A) [20,21]. This may influence HBV replication by abnormally regulating various cellular processes such as DNA repair, cell growth, differentiation, adhesion, and proliferation, which may promote hepatocarcinogenesis [10,15,22-24].
HBx and transforming growth factor beta 1
Transforming growth factor beta (TGF-β), encoded by TGF-β gene, is required for wound healing and hepatic tissue repair. It regulates several cellular functions including cell growth, differentiation, apoptosis and homoeostasis [25]. TGF-β belongs to the TGF super family that includes various isoforms: TGF-β1, TGF-β2 and TGF-β3 [26]. TGF-β1 is a cytokine that is produced in response to liver injury by activated hepatocytes, platelets and Kupffer cells [27]. As a transcriptional transactivator, HBx protein suppresses TGF-β-induced apoptosis through activation of PI3K pathway that contributes to hepatocarcinogenesis by cross-talking with other pathways such as AKT/mTOR and Ras/MAPK (Figure 1A) [28]. HBx-mediated upregulation of TGF-β1 and downregulation of α2- macroglobulin promotes proliferated hepatic stellate cells leading to HBV-related fibrosis [11,27,29]. Upregulation of TGF-β1 correlates with the mutation and loss of mannose-6-phosphate/IGF-II receptor that mediates TGF-β1 activation leading to HBV-induced HCC [30-32]. Liu and co-authors have shown that HBV promotes hepatocarcinogenesis in BALB/c mice through upregulation of SMAD7 and inhibition of TGF- β-induced apoptosis [33]. In human hepatic stellate cells, upregulation of TGF-β1 and its downstream mediator of fibrogenic action known as connective tissue growth factor lead to enhanced cell proliferation and progressive fibrosis [34].
HBx and vascular endothelial growth factor
HCC was recently labelled a hypervascular tumour due to its association with vascular endothelial growth factor (VEGF)-mediated activities that promotes vasculogenesis and angiogenesis [35]. Activation of COX-2-mediated PGE2 enhances the expression of VEGF and tumour angiogenesis in HBV-related HCC [36]. In dysplastic nodules of hepatocarcinogenesis, upregulation of VEGF-A and its co-operating receptors FIK-1 and hypoxia-inducible factor-1 alpha (HIF-1α) promote angiogenesis and support sustained growth of these precursor lesions contributing to the formation of hepatic cancer and metastases [37]. Production of VEGF occurs via activation of several pathways including mTOR, IκB kinase β (IKKβ), NF-κB, ribosomal protein S6 kinase 1 and Rac (Figures 1A and 1B) [37-41]. HBx-induced expression of VEGFR-3 splice variant in HCC patients correlates with tumour aggressiveness, tumour relapse and poor prognosis [42].
HBx and interleukin-8
Interleukin-8 (IL-8) is a leukocyte chemotactic activating cytokine secreted in response to an inflammatory stimulus by macrophages, endothelial and epithelial cells. IL-8 may function as a regulatory factor within the tumour environment and it is implicated in various cellular signalling including cell growth, proliferation, angiogenesis and migration. Upregulation of IL-8 in chronic HBV infection has been observed, and it was found to correlate with interferon-alpha therapy resistance, advanced liver inflammation and fibrosis [43,44]. Previous studies have shown that HBx protein increases the expression of IL-8 by interacting with NF-κB and CCAAT enhancer-binding protein (C/EBP)-like cis elements [45]. C/ EBP-like cis element regulates the expression of COX-2, another protein implicated in HBx transcriptional transactivation activity. Overexpression of COX-2 significantly correlates with increased HBx protein in HCC, suggesting that COX-2 may be hijacked in influencing HCC-related micro angiogenesis and metastases [46]. This may be explained by the co-operative network and triple effects of IL-8, IL-29 and COX-2 when upregulated in response to increased viral replication in chronic HBVinfected patients and hepatoma cultured cells. It appears that HBV induces a differential regulatory network of inflammatory responses in which IL-29, IL-8 and COX-2 regulate one another. In this way, upregulation of IL-29 by HBV activates IL-8 that in turn suppresses IL-29 production (Figure 1B). This enhances the translocation of cAMP response element binding (CREB) and C/EBP transcription factors from cytosol to nucleus by stimulating ERK and JNK signalling pathways, which activate COX- 2 and PGE2 production leading to enhanced HBV replication associated with severe inflammation and tumorigenesis. COX-2 also represses the production of IL-8, and IL-29 induces antiviral factors protein kinase R and 2’-5’ oligoadenylate synthetase leading to suppressed HBV replication [47].
HBx and p53 protein
HBx protein contributes to hepatocarcinogenesis by blocking p53- mediated cellular processes that are important for maintaining the genomic integrity of hepatocytes [48,49]. In normal circumstances, p53 regulates apoptosis by interacting with cytoplasmic transcription factors such as repair cross-complement in grodent-repair deficiency group 2, xanthoma pigmentosa B, fatty acid synthase, p21CIPWARF1, and ankyrinrepeat containing and proline-rich region-containing proteins (ASPP). These transcription factors are implicated in the p53-mediated nucleotide excision repair and enhance the binding of p53 gene to proapoptotic stimuli [13,50-52]. Suppression of p53 protein-mediated apoptosis also occurs via activation of cyclooxygenase 2 (COX-2)-prostaglandin E2 and McI-1 anti-apoptotic pathways (Figure 1C). In cultured hepatic oval cells, HBx protein promotes cell proliferation by enhancing the expression of Let-7a-microRNA, signal transducer and activator of transcription 3 (STAT3) and cyclin D1 though activation of MAPK/ERK and P13Kdependent signalling pathways [24]. Interaction of HBx protein and p53 mutant correlates with progressive tumour formation driven by the activation of MYC, JNK, VEGF and phosphatase and tensin homolog (PTEN) through PI3K/AKT pathway [53]. Inhibiting the transcription activities of HBx protein could lead to suppressed tumour initiation, growth and metastases.
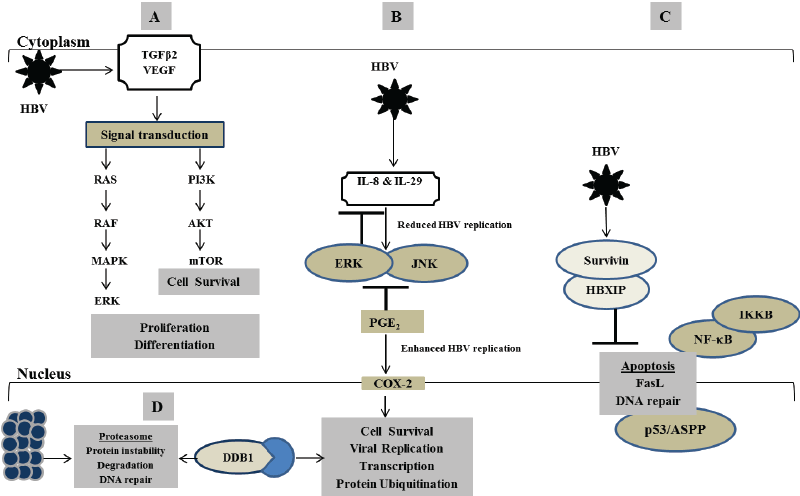
Figure 1: Mechanisms underlying how HBX protein transcriptionally transactivates various cellular genes leading to disruption in multiple biological processes that are critical for the development of HBV-induced hepatocellular carcinoma. (A) Production of TGFβ2 and VEGF via activation of several pathways including PI3K, mTOR, IκB kinase β (IKKβ), NF-κB, ribosomal protein S6 kinase 1 and Rac. (B) Regulation of IL-8 and IL-29 through ERK and JNK signalling pathways. (C) Suppression of p53 protein-mediated apoptosis via activation of HBx-survivin-HBXIP complex. (D) Induction of apoptosis or cell proliferation associated with aberrant chromosome segregation through interaction of HBx and DDB1 protein
HBx protein and survivin-HBXIP complex
Several studies demonstrate that anti-apoptotic protein survivin forms a complex with hepatitis B-X interacting protein (HBXIP), and that this complex interacts with HBx protein in contributing to hepatocarcinogenesis. Survivin is an anti-apoptosis gene expressed in various human malignancies, and its upregulation is implicated in HBxassociated HCC [54-56]. HBXIP is a conserved 18 kDa ubiquitous protein that was first discovered as a binding partner for HBx protein, and it negatively regulates HBx protein activity leading to disruption in the HBV replicative cycle [56]. Mouse studies have shown that HBXIP is required for hepatocyte growth and survival; it functions as a binding partner for survivin [57]. HBx protein interacts with survivin-HBXIP complex and correlates with dysregulated centromere dynamics and mitotic spindle formation (Figure 1C) [58]. This interaction was found to supress caspase activation, modulate p53 checkpoints and control spindle formation and proper kinetochore attachment cell division in a survivin-dependent manner that exacerbates hepatocarcinogenesis. HBx protein interacts with survivin-HBXIP complex and promotes cell cycle arrest and suppressed hepatoma cell growth through mechanisms that modulate oncoprotein HBXIP and tumour suppressor miR-520b [57,59,60]. Hu and co-authors [61] have shown that miR-520b impedes breast cancer cell migration by targeting andregulating the expression of IL-8 and HBXIP. Following this line of evidence, deactivation of mitogen-activated protein kinase kinase 2 (MEKK2) and cyclin D1 resulted in the downregulation of miR-520b and hepatoma cell proliferation in HCC, suggesting potential therapeutic targets [62]. In contrast, HBx protein upregulates MEKK2 through activation of transcription factor AP-2 alpha (TFAP2A) and sphingosine kinase 1 (SPHK1), and this leads to tumour aggressiveness [63,64]. MEKK2 is a member of the MAPK signalling pathway that activates the JNK/MAPK pathway and ERK5 leading to regulation in tumour growth and metastasis [64].
HBx and damage-specific DNA binding protein 1
Damage-specific DNA binding protein 1 (DDB1) is the wellcharacterised binding partner of HBx protein that may contribute towards promoting viral replication, and their interaction was found to be conserved in all mammalian hepadnaviruses including woodchucks [65,66]. DDB1 is a 127kDa protein that binds to DDB2, a protein that facilitates its transportation in the nucleus. This binding forms a heterodimeric DNAdamage binding complex that functions in nucleotide-excision repair pathway and recognises the DDB2 ultraviolet-induced DNA damage. DDB1 also serves as an adaptor for the culin 4 (CUL4)-DDB1 ubiquitin E3 ligase complex that ubiquitinates and degrades substrate proteins by the proteasome contributing to hepatocarcinogenesis [67,68]. Although they enter the nuclear compartment separately, HBx protein interacts with DDB1 to activate viral replication by interfering with hepatocyte viability in cell culture. Interaction of HBx and DDB1 protein was thought to interfere with CUL4-DDB1 ubiquitin E3 ligase complex and cause genome instability by inducing apoptosis or cell proliferation associated with aberrant chromosome segregation (Figure 1D) [69,70]. However, a recent study has shown that there were no differences in the levels of HBV DNA replication in cells transfected with DDB1 expression plasmid carrying a wild-type HBx replicon as compared to the one with X-null HBx replicon [71]. This study suggested that DDB1 interacts with HBV covalently closed circular DNA (cccDNA) and promotes viral replication via an unknown mechanism that does not involve interaction with HBx protein. Interaction of DDB1 with DDB2 could also induce transcriptional activation either by being directly recruited to the cccDNA by the p300/ CBP histone acetyltransferase (HAT) or by acting as a transcriptional factor that interacts with E2F1 and Sp1 transcription factors which disrupt cell cycle control [72-74].
Effects of HBx-induced Epigenetic Alterations in Hepatocarcinogenesis
Epigenetics is a non-mutational alteration of gene expression that occurs through epigenetic marks or tags such as methylation of DNA and covalent modification of histones proteins [75,76]. DNA methylation attaches the methyl groups to the nucleotide sequence via catalysis by several DNA methyltransferases (DNMTs). Histone modifications either add or remove chemical groups to or from histones via histone modifying enzymes such as HATs and histone deacetylases (HDACs). HBx protein has been shown to trigger aberrant epigenetic signatures that influence HBV-induced hepatocarcinogenesis [77-79]. However, the precise mechanisms of action are still being elucidated.
Tumour suppressor genes are often aberrantly repressed due to DNA hypermethylation. This type of epigenetic lesion describes an addition of methyl group in the 5’-methylcytosine of gene promoter regions [80]. HBx gene has been repeatedly reported to be frequently integrated and preferentially maintained in patients with HBV-related HCC. HBx protein induces the hypermethylation of several tumour suppressor genes by modulating the transcriptional activation of DNMTs that result in the loss of gene expression and normal functions leading to hepatocarcinogenesis [81-88]. Several promoter regions encoding ASPP, retinoic acid receptor β2 (RARβ2), insulin-like growth factor binding 3 (IGFB3), caveolin- 1(Cav1), long interspersed nuclear elements-1(LINE1), retinoblastoma (pRB), E-cadherin, glutathione S-transferase P1(GSTP1) and human telomerase reverse transcriptase (hTERT) tumour suppressor genes have been shown to be repressed via HBx-induced DNMT1 and DNMT3A hypermethylation (Figure 2) [81-88]. HBx-induced hypermethylation may disrupt cellular signalling pathways such as ubiquitination, DNA repair, transcription, proliferation and apoptosis accompanied by tumour development, aggressiveness and metastases (Figure 2) [81-88]. HBxmediated downregulation of secreted frizzled-related protein (SFRP)- 1 and SFRP5 in HBV-related HCC tissues was significantly associated with upregulation of DNMT1 that led to poor tumour differentiation by disrupting Wnt pathway. Silencing the expression of DNMT1 with methylation inhibitor restored SFRP1 and SFRP5 expression leading to inhibition in HCC growth and regression of HBx-induced EMT [89]. DNA hypomethylation signifies the loss of methylation that affects mostly repeated sequences and it is accountable for the global DNA hypomethylation that is frequently observed in several malignancies. In contrast to DNA hypermethylation, hypomethylation correlates with activation of proto-oncogenes. Upregulation of insulin-like growth factor 2 (IGF2) oncogene via HBx-induced hypermethylation coincides with poor clinical outcome in HCC patients [90].
DNA methylation can collaborate with histone modifications and alters hepatic gene expression synergistically. Silence in DLEC 1 gene expression is mediated by both DNA hypermethylation and histone acetylation [86]. HBx-induced expression of deleted in lung and esophageal cancer 1 (DLEC 1) gene via activation of HATs leads to suppression of tumour progression [91]. Through the activation of DNMT1 expression mediated by the pRB-E2F pathway, HBx protein induces DNA hypermethylation of DLEC 1 gene and suppresses its transcriptional activities [91]. HBx protein also allows the establishment of active chromatin by interacting with lysine specific histone demethylase 1 (KDM1A) and su(var) 3-9, enhancer of zeste, trithorax 1A (SET1A) enzymes which trimethylate H3K4, an epigenetic mark associated with active transcription [92]. HBV cccDNA is tightly packed in the nucleus as an episomal DNA, and it is required for viral persistence and replication. HBV cccDNA is packaged into minichromosomes by histone and non-histone proteins [93,94]. It has recently been shown that HBx protein activates SET domain bifurcated 1 and recruits heterochromatin protein 1 leading to silence in the transcription of HBV cccDNA via trimethylation of H3 on lysine 9 (H3K9me3) [95]. It is evident that HBx protein regulates HBV replication and transcription by remodelling minichromosomes.
Cooperative networking between DNA methylation and noncoding miRNA (miR) in regulating gene expression and promoting hepatocarcinogenesis has also been repeatedly reported [62,96]. HBx protein promotes hepatocarcinogenesis by inducing hypermethylation via activation of DNMTs and silencing gene transcription in the promoter region of tumour suppressor genes such as miR-21, miR-101, miR-152 and miR-205 (Figure 2) [61,62,97,98]. Suppressed expression of miR- 205 was associated with HBx-enhanced hepatocyte transformation and proliferation that favour malignant transformation and subsequently cancer development [99]. Upregulation of HBx-induced miR-21 leads to the loss of programmed cell death 4 gene expression and normal function in HCC patients, providing another novel insight into mechanisms underlying HBV-related HCC pathogenesis [96].
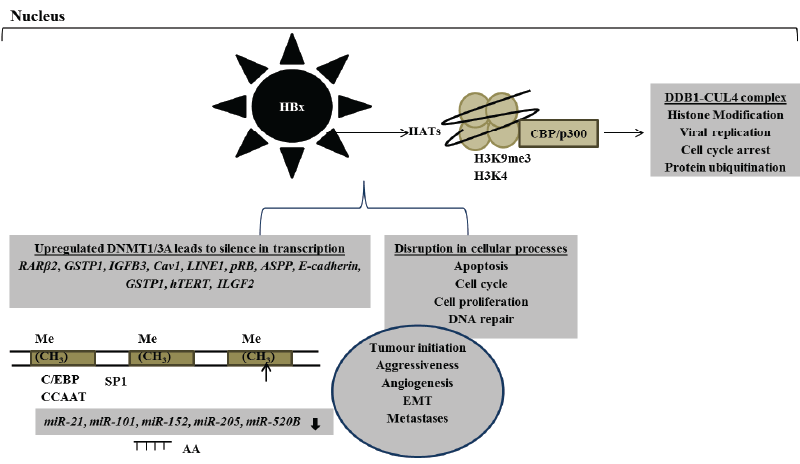
Figure 2: Aberrant HBx-induced epigenetic mechanisms involved in the alteration of gene transcription and normal cellular processes that are important in hepatocarcinogenesis
Summary
As a multifunctional protein, HBx exerts its actions by either interacting with key transcriptional factors or epigenetically regulating tumour suppressor genes that are critical for HBV-related hepatocarcinogenesis and metastases. Current literature shows HBx protein triggers epigenetic abnormalities and disrupts cellular signalling pathways that favour uncontrolled hepatocyte proliferation, development of HBV-induced inflammation, fibrosis and cancer. Despite the reported evidence, the role of HBx protein in the pathogenesis of HBV-related HCC still remains enigmatic and therefore warrants further investigation that will characterise the structure and functions of this protein. This will provide insight into molecular mechanisms underlying the role of HBx protein as an epigenetic and gene transcription regulator.
Acknowledgement
This work was supported by the National Research Foundation, Medical Research Foundation and Oppenheimer Memorial Trust.
Conflict of Interest
The authors declare no conflict of interest.
Article Information
Aritcle Type: Review Article
Citation: Kgatle MM, Kalla AA, Spearman CW, Hairwadzi HN (2016) The Role of HBx-mediated Transcriptional Activities and Epigenetic Alterations in Hepatitis B Virus-induced Hepatocellular Carcinoma. J Emerg Dis Virol 3(1): doi http://dx.doi. org/10.16966/2473-1846.126
Copyright: © 2016 Kgatle MM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 14 Nov 2016
Accepted date: 12 Dec 2016
Published date: 16 Dec 2016