Case Study
The patient is a 12 year old Caucasian boy who presented to his primary care physician with h/o fatigue, lethargy, low grade fevers and vomiting. His past medical history was significant for primary nocturnal enuresis and constipation. For the enuresis patient has seen an urologist and was started on desmopressin about 4 years back. Prior lab works included a urine analysis which showed low specific gravity at 1.005 but no protein or blood. For constipation, patient uses stool softener on and off. No other significant health issues were identified by family. He was one of four siblings and there was no significant family h/o renal disease, dialysis or kidney transplant. The pediatrician did lab work including a complete blood count and chemistry panel which showed the following values. Hemoglobin 8.2 g/dl, Hematocrit 23%. Sodium 140 meq/L, potassium 4 meq/L, chloride 109 meq/L, bicarbonate 20 meq/L , blood urea nitrogen 90 mg/dl, creatinine 4.1 mg/dl, calcium 8.1 mg/dl and phosphorus 6.1 mg/dl.
Patient got admitted under pediatric nephrology service in Children’s hospital for further evaluation. Physical examination showed a height of 146 cm, body weight of 34 kg, and blood pressure of 114/66 mm Hg with a regular heart rate of 90 beats per minute. On exam no edema was appreciated. Visual acuity was normal and an ophthalmologist assessment did not show any retinal abnormalities. Neurological exam was intact and no skin rash or joint swelling appreciated. Further work up included complements (C3 & C4) which were in the normal limits, Parathyroid hormone elevated at 966 pg/ml indicating chronic kidney damage and vitamin D level was adequate at 27 ng/ml. Urine analysis was repeated and showed bland urine with low specific gravity 1.005 and no protein or blood. Urine protein/Creatinine ratio was normal at 0.2 and fractional excretion of sodium was elevated at 7.6% indicating intrinsic renal injury.
Imaging studies included a renal ultrasound which showed both kidneys to measure around 10 cm (normal for age) with increased echogenicity and no hydronephrosis. A voiding cysto urethrogram (VCUG) did not reveal any reflux. Radionuclide studies included a MAG3 scan which showed slow uptake of the radiotracer with cortical retention and slow delayed excretion compatible with chronic medical renal disease. A clinical diagnosis of cystic kidney disease was made and molecular testing for NPHP 1 gene done. Patient’s acidosis was corrected with bicarbonate supplementation and epogen was started for anemia. The molecular testing did come back positive for NPHP1 homozygous deletion confirming the diagnosis of nephronophthisis. Patient was placed on a preemptive renal transplant list and is now s/p a diseased donor kidney transplant with normal renal function. Screening on his other siblings showed that one of his younger brothers, an eight year old boy also has NPHP with elevated creatinine and is currently getting medically managed for his chronic kidney disease.
Background
Nephronophthisis (NPHP) is an autosomal recessive condition leading to cystic kidney disease and is a leading genetic cause of renal failure in young children and adolescents [1]. The incidence of NPHP varies worldwide from 1 in 50,000 to 1 in 900,000 children with a reported prevalence of 5% among pediatric end stage renal disease (ESRD) patients in the United States [2]. Because of shared morphological features, autosomal dominant medullary cystic kidney disease (MCKD) and NPHP are often described together. The key difference between NPHP and MCKD is age of onset. The median age of ESRD due to NPHP is 13 years, while MCKD usually progresses to ESRD in adulthood.
NPHP is a genetically heterogeneous disease with 13 identified genetic mutations accounting for 30% of all affected patients [3]. The protein products of most of the mutated genes localize to the primary cilium in accordance with the concept of ciliopathies. Infantile NPHP has been linked to NPHP2 mutations, while the more common juvenile form has mutations in several genes including NPHP 1, 4, 5 and 6. Mutation in Nephrocystin-1 (NPHP1) accounts for majority of isolated cases of NPHP [4,5].
A variety of extra renal manifestation can occur with NPHP including retinitis pigmentosa, oculo motor apraxia, cerebellar vermis hypoplasia, occipital encephalocele, coloboma of the optic nerve, Leber congenital amaurosis (LCA), hepatic fibrosis and situs inversus illustrating the multitude of downstream effects of ciliopathies. A brief description of the syndromes associated with NPHP and extra renal features common in those syndromes is described below.
Bardet-Biedl syndrome (BBS)
BBS is a rare autosomal recessive ciliopathy with incidence ranging from 1 in 140,000 live births in North America to1 in 13,500 live births in Kuwait, where consanguinity is more common [6]. Sixteen different BBS gene mutations have been identified, with the predominate genotypes of disease being BBS1 and BBS10 [7]. Clinical features involve multiple organs and include postaxial polydactyly and progressive loss of vision due to rod cone dystrophy [8]. Obesity is present in majority of patients with one third of patients becoming obese by one year of age [9]. Hypogonadism and infertility are usually present. Renal involvement contributes to the majority of mortality and morbidity in these patients and can present with varying degrees of dysplasia and cystic disease.
The varying gene products in BBS localize near the basal body of the primary cilium and are involved in the formation of BBSome and chaperonin complex. These in turn interact with BBS3 (GTPase protein) and rab8 facilitating formation and maintenance of cilia [10].
Joubert syndrome and Joubert syndrome related disease (JS, JSRD)
JS is an autosomal recessive inherited condition with incidence ranging from 1 in 80,000 to 1 in 100,000 live births; and is characterized by the hallmark finding of “molar tooth sign” on brain imaging secondary to a complex midbrain-hindbrain malformation [11,12]. The neurological features include varying degrees of hypotonia, ataxia, developmental delay, oculomotor apraxia, nystagmus and neonatal breathing dysregulation with alternating episodes of hyperpnea and apnea. As with the clinical features of other ciliopathies, multi- organ involvement mainly retinal dystrophy, LCA, nephronophthisis, hepatic fibrosis and polydactyly are often present. Renal involvement is present in about 25% of patients with JS.
Ten causative genes have been identified (JBTS 1 to 10) which localize to the primary cilia. Genetic and clinical overlap with other syndromes especially Meckel Gruber syndrome (MKS) is seen. Genetic mutations which contribute to the majority of JSRD and their associated clinical features are presented in table 1.
JBTS |
Mutated protein |
Genetic locus |
Localization |
Clinical associations |
JBTS3/AHI1 |
Jouberin |
6q23.3 |
Primary cilia |
RD, PM, later presentation of renal impairment |
JBTS4/NPHP1 |
Nephrocystin-1 |
2q13 |
Transition zone/base of primary cilia |
NPHP, RD, OMA, MTS |
JBTS5/CEP290 |
Centrosomal protein 290 |
12q21.32 |
Primary cilia, centrosome, retinal photoreceptor connecting cilium |
RD, NPHP, LCA, encephalocele |
Table 1: Genetic defect in JSRD [11]
RD: Retinal dystrophy; PM: Polymicrogyria; NPHP: Nephronophthisis; OMA: Oculo motor apraxia; LCA: Leber congenital amaurosis
Senior-loken syndrome (SLS)
The association of NPHP with retinal degeneration is referred to as SLS, which can occur independently or as a part of JS. Two variants of retinal disorder namely Leber congenital amaurosis (LCA) and tapetoretinal degeneration can occur with SLS [13]. Leber congenital amaurosis is the most severe variant and can present with profound loss of vision, nystagmus with varying degrees of atrophy and pigmentary changes in the retina. Tapetoretinal degeneration is a milder variant and it presents with tube like restriction of visual field and varying degree of pigmentary alteration in the retinal field.
Meckel gruber syndrome (MKS)
MKS is a lethal autosomal recessive disease with incidence ranging from 1 in 13,250 to 1 in 140,000 live births with predilection for Belgian and Finnish populations [14]. It is characterized by a triad of malformations, usually occipital encephalocele, cystic dysplasia of the kidneys and post axial polydactyly [15,16]. Six different genes have been identified up to date (MKS1 to 6) which localize to the primary cilia and play a role in the structure and proper function of cilia [17].
Boichis syndrome and RYHNS syndrome
The association of nephronophthisis with congenital hepatic fibrosis has been described as Boichis syndrome [18]. Association of NPHP with varying degree of retinal involvement, skeletal dysplasia and pituitary deficiency is called the RHYNS (Retinitis pigmentosa, Hypopituitarism, Nephronophthisis and Skeletal dysplasia) syndrome [19].
Cogan syndrome
Cogan syndrome is an autosomal recessive condition which presents with oculomotor apraxia with defective horizontal eye movements and nystagmus. Patients with Cogan syndrome can have cerebellar vermis hypoplasia [20]. Deletions and point mutations in NPHP 1 gene of patients with Cogan syndrome can have cerebellar vermis hypoplasia as well [21].
Jeune syndrome or Asphyxiating Thoracic Dystrophy (ATD)
Jeune syndrome or ATD is an autosomal recessive osteochondrodysplasia with characteristic skeletal abnormalities including narrow thorax, short ribs, short bones in arms and legs, polydactyly and short stature [22]. Varying degrees of renal, hepatic, pancreatic and retinal complications occur in children who survive beyond the first few years of life. Renal involvement occurs in about 30% of patients and includes varying degrees of cystic dysplasia, hypertension and progressive renal insufficiency. Genes mutated in Jeune syndrome (IFT80 and DYNCH1) play a main role in intra flagellar transport and hence essential for maintenance of ciliary structure and function.
Genetic basis of NPHP
A growing number of genes have been implicated in NPHP, inherited in an autosomal recessive manner (Table 2). Oligogenicity, in which allelic variants at multiple locations can contribute to the disease, and epistasis in which modifier genes can alter phenotype, have been identified with NPHP [23,24]. Oligogenicity and epistasis explains the wide spectrum of clinical variation that can be associated with any mutant gene in NPHP.
NPHP type |
Mutated protein |
Localization |
Extra renal manifestation |
NPHP 1 |
Nephrocystin- 1 |
Transition zone/base of primary cilia |
SLS, JS |
NPHP 2 |
Inversin |
Primary cilia |
SLS, Situs inversus, Hepatic fibrosis |
NPHP 3 |
Nephrocystin-3 |
Primary cilia (adherens junctions) |
SLS, MKS, Situs inversus, Hepatic fibrosis |
NPHP 4 |
Nephrocystin-4/nephroretinin |
Primary cilia (adherens junctions) |
SLS |
NPHP 5 |
Nephrocystin -5 |
Primary cilia (adherens junctions) |
SLS |
NPHP 6 |
Centrosomal protein 290 |
Primary cilia |
LCA, MKS, BBS, SLS, JS |
NPHP 7 |
Gli similar protein 2 |
Primary cilia (nucleus) |
|
NPHP 8 |
RPGRIP1-like |
Primary cilia (Basal body and centrosome) |
SLS, JS, MKS |
NPHP 9 |
Never in mitosis A-related kinase 8 |
Primary cilia (Basal body and centrosome) |
SLS |
NPHP 10 |
Serologically defined colon cancer antigen 8 |
Primary cilia (centrosome and cell cell junction) |
SLS, BBS |
NPHP 11 |
Transmembrane protein 67 |
Membrane of primary cilia |
JS, MKS, Hepatic fibrosis |
Table 2: Genetic defect and mutated protein in NPHP
SLS: Senior Loken syndrome; JS: Joubert syndrome; MKS: Meckel Gruber syndrome; LCA: Leber Congenial amaurosis; BBS: Bardet Biedl Syndrome
NPHP1: NPHP1, which accounts for approximately 25% of nephronophthisis cases, was the first gene identified to cause this group of diseases [25]. Most common among NPHP1 mutation is homozygous deletion at 2q13 [26]. Most common extra renal manifestation with NPHP1 mutation includes SLS, JSRD and Cogan syndrome [27-29]. The protein product of NPHP1, namely nephrocystin-1, is expressed predominantly in the renal collecting ducts [30] and localizes to the primary cilium and epithelial cell adherens junctions [31,32].
NPHP2: Mutations in NPHP2 give rise to infantile NPHP and account for <1% of cases [33]. NPHP2/INV is located in 9q31.The protein product inversin is located in the primary cilium and other subcellular sites dynamically based on the stage of cell cycle [34]. Extra renal manifestations include situs inversus, ventricular septal defect, hepatic fibrosis and rarely SLS [35]. In addition to its proposed mechanism of acting as a switch between canonical and non-canonical Wnt pathway, inversin plays a role in maintenance of tubular architecture via planar cell polarity signaling [36].
NPHP3: NPHP3 mutations are rare, accounting for <1% of cases. A variety of extra renal phenotype including SLS, MKS and situs inversus can be present. NPHP3 gene encodes for protein nephrocystin-3 and is located in 3q22.1 [37]. Nephrocystin-3 localizes to the primary cilium and interacts with both nephrocystin-1 and inversin [38].
NPHP4: NPHP4 which encodes nephrocystin-4 or nephroretinin has a mutation frequency of 2 to 3% in genetically confirmed NPHP with the abnormality located in 1p36.22 [39,40]. Nephrocystin -4 localizes to the primary cilium and interacts with other proteins namely nephrocystin 1,3,8 and inversin. Retinitis pigmentosa is the most common extra renal phenotype associated with NPHP4 mutation explaining the name nephroretinin and its association with retinal ciliopathy gene retinitis pigmentosa GTPase regulator (RPGR).
NPHP5: Mutations in NPHP5 affect nephrocystin-5, which contains two IQ calmodulin binding sites located in 3q21.1 [41]. NPHP5 mutations cause early onset retinal degeneration. Nephrocystin-5 colocalizes with nephrocystin-1 and nephrocystin -4 in the primary cilium. Nephrocyctin-5 is similar to nephroretinin in that it complexes with RPGR explaining the retinal involvement.
NPHP6/CEP290: NPHP6 also known as CentrosomalProtein 290 (CEP290) is located in 12 q21.32 [42]. Mutations in NPHP6 account for differing clinical phenotypes including isolated NPHP, SLS, JS, MKS and BBS. NPHP6 is also the most common isolated mutation seen in Leber’s congenital amaurosis [43]. The varying clinical phenotypes with NPHP6 mutations have been suggested to be secondary to its oligogenicity. NPHP6 also interacts with other transcription factors such ATF4 which is involved in renal cyst formation and coiled-coil and c2 domain protein (CC2DA) [44]. CC2DA mutations have been noted in JS and MKS [45].
NPHP7/GLIS2: NPHP7 encodes GLI similar 2 protein (GLIS2) and is located in 16 p 13.3 [46]. GLIS2 localizes to the primary cilium and nucleus.
NPHP8/RPGRIP1L: NPHP8 encodes retinitis pigmentosa GTPase regulator interacting protein 1-like (RPGRIP1L) and is located in 16q12.2 [47]. NPHP8 mutations often cause extra renal manifestations such as JS, MKS and cerbello-oculo-renal syndrome. RPGRIP1L co-localizes with nephrocystin-4 and 6 at the basal bodies and centrosomes [48].
NPHP9/NEK8: NPHP9 encodes NIMA-kinase 8 (NEK8) protein and is located in 17 q11.1 [49]. NEK8 co-localizes with various nephrocystins in primary cilia and also has shown interaction with polycystin-2 [50]. Although a rare cause for NPHP, NPHP9 mutation has been shown to cause both infantile and non-infantile NPHP.
NPHP10/SDCCAG8: NPHP10 encodes Serologically Defined Colon Cancer Antigen 8 (SDCCAG8) and is located in 1 q44 [51]. SDCCAG8 colocalizes with nephrocystin-5, encoded by NPHP5, in centrosomes and the cell-cell junction of the primary cilium. It also interacts with retinal ciliopathy proteins such as RPGRIP1L, encoded by NPHP8. Though a rare cause for isolated NPHP, SDCCAG8 mutations have been noted in SLS and BBS.
NPHP11/TMEM67: NPHP11 encodes trans-membrane protein 67 (TMEM67) and is located in 8 q22.1 [52]. NPHP11 mutations have been noted in patients with NPHP and liver fibrosis and other ciliopathies such as JS and MKS. TMEM67 localizes to the primary ciliary membrane and plays an important role in maintaining ciliary cellular structure.
Mutations have also been identified in protein products other than ciliary proteins such as X-propyl aminopeptidase (XPNPEP3) which localize to mitochondria in two consanguineous families with NPHP [53]. Additional extra renal manifestations noted in the affected family include cardiomyopathy and seizures. Another rare mutation linked to retrograde intraflagellar transport (IFT139) has also been identified in some families with NPHP [54].
Role of Cilia in Cystic Kidney Disease
Cilia are finger like projections from the surface of cell with a modified cell membrane. Cilia are microtubule based structures that grow out from basal bodies or centrosomes [55,56]. Depending on the microtubule cytoskeleton, cilia can be classified into primary or immotile or nonflagellated and secondary or motile or flagellated. Motile cilia found in the respiratory and reproductive tracts contains 9 peripheral doublets with 2 central microtubules (9+2 arrangement) while the primary ones have no central microtubule (9+0 arrangement). Primary cilia are highly conserved organelles through evolution and are present in almost all the vertebrate cell types except for lymphocytes and intercalated cells of the distal renal tubule. Primary cilia carry out different functions based on cell type. They can detect a variety of mechanical, osmotic, photonic and olfactory stimuli and play a role in controlling cell cycle and polarity of epithelial cells.
Primary cilia assemble and disassemble at various stages of the cell cycle. Intraflagellar transport is mediated by transport proteins such as kinesin-2 which promotes anterograde movement while dynein directs retrograde movement. Disruption of transport in either direction will interfere with cilia formation and function [57-59]. As genes involved in varying cystic kidney diseases were identified with positional cloning, it was observed that the mutated proteins localize to varying parts of primary cilia, centrosome and basal bodies [60]. This lead to evolution of a new disease concept, namely ciliopathy, thus linking the role of malformed and malfunctioning cilia to renal cystic disease. The proposed mechanisms linking cilia and cyst formation can be classified into the following;
1. Intact cilia and polycystin 1 and 2 are shown to be essential in flow induced calcium release from the endoplasmic reticulum in cultured Madin-Darby canine kidney cells through ryanodine and inosine triphosphate (IP3) receptors [61-63]. This indicates the role of polycystins as a sensory modulator in cilia. Lower intracellular calcium has been postulated to reduce clearance of intracellular cAMP which in turn can cause increased cell proliferation and abnormal fluid secretion leading to cyst formation [64].
2. The role of mammalian target of rapamycin (mTOR) in cyst formation came to light with identification of tuberous sclerosis genes (TSC1 and TSC2) which codes for protein tuberin and hamartin respectively. TSC1 like polycystin also localizes to the cilium [65-67]. TSC1 and TSC2 forms a complex with a small GTP binding protein namely Rheb and this in turn is inhibited by phosphatidyl inositol 3 kinase/protein kinase B (PI3K/ Akt or PAkt) signaling. The mTOR complex (mTORC1) which activates variety of cellular processes such as growth and proliferation is inhibited by TSC1/TSC2/Rheb complex while phosphorylation of TSC2 by PAkt signaling will indirectly activate mTORC1. Uninhibited mTORC activity could result in reduced apoptosis and abnormal cell proliferation and subsequent formation of cyst.
3. Primary cilia and the cystoproteins play a main role in Wnt signaling pathways, a deregulation of which can contribute to cyst formation. Wnt signaling pathways are classified into canonical and non-canonical pathways. The initiation process for Wnt signaling involves interaction of Wnt ligands with specific Frizzled (Fz) receptors which recruits intracellular Dishevelled proteins (Dvl) and further co-receptor activation [68,69]. The canonical pathway is β-catenin dependent while non canonical pathway is β-catenin independent and determines planar cell polarity (PCP).
Overexpression of β-catenin has shown to cause cyst formation due to defective cell turnover and abnormal ion channel localization in animal models [70]. Abnormalities in Wnt/PCP signaling pathway has shown to cause abnormal polarity in tubular epithelia cells, disordered orientation of cell division ultimately causing abnormal tubular elongation and cyst formation [71,72].
4. Inversin, a protein product of NPHP 2 is located in the primary cilium. Inversin acts as a switch between canonical and non-canonical pathway. Absence of this switch due to mutation in NPHP2 can cause sustained canonical pathway and cause abnormal cell proliferation and division.
5. Cilia also play a role in hedgehog (Hh) signal transduction. Hh signaling involves binding of three morphogens namely sonic (Shh), Indian (Ihh) and Desert (Dhh) to its receptor Patched 1 (Ptch1). The ligand receptor interaction derepresses and activates trascriptional factors such as Gli 2 and Gli 3 [73,74]. Mutations in ciliary proteins can cause abnormal Hh activity and can lead to abnormal renal development. Reduced Hh signaling has been shown to be associated with ectopic kidney and cyst formation in mice [75] while Ihh has been found to be upregulated in a corticosteroid induced models of renal cyst [76].
Evaluation and Screening Strategies
Early presenting features in NPHP are usually subtle and are secondary to impaired concentrating ability of the kidneys. Initial presentation often includes polyuria, nocturia, polydipsia and secondary enuresis. Anemia and lethargy presents early in the disease. Early morning urine will be inappropriately dilute due to the inability to concentrate in the setting of water restriction [77]. Gradual deterioration of renal function occurs with progression to ESRD by adolescence in juvenile NPHP. In the infantile form children may reach renal failure by 3 years of age.
Diagnosis of NPHP relies on a clinical suspicion of the disease. Cystic kidney diseases should be considered in the differential in children presenting with polyuria, polydipsia, enuresis and low urine concentrating ability. Other clinical presentations could include but not limited to complications of renal insufficiency such as nausea, vomiting, fatigue due to anemia and pruritus due to uremia. Blood pressures may be elevated but likely normal in the initial stages secondary to polyuria. Presence of extra renal manifestations such as abnormal eye movements, abnormal retinal pigmentation, polydactyly and other neurological manifestations along with a family history of renal disease or consanguinity should alert the clinician to the possibility of a ciliopathy.
Initial investigation would include measurement of first morning urine osmolality and protein/creatinine ratio. Typically proteinuria is minimal or absent and the urine sediment is bland. Low morning urine osmolality indicates defect in urine concentrating ability. Renal function, liver function, complete blood count, and intact parathyroid hormone give essential information in diagnosis and management of chronic kidney disease. An ultrasound of the abdomen will show normal or reduced kidney size usually with increased echogenicity. Cortico-medullary cysts are often present but sometimes may not be sonographically apparent during earlier stages of the disease.
A baseline ophthalmological examination is recommended to evaluate retinal and ophthalmological changes associated with NPHP. Depending on the clinical presentation further neurological imaging and evaluation may be necessary. Ongoing surveillance of growth and development, endocrine function, and sexual maturation in the setting of chronic kidney disease is essential.
Because NPHP1 accounts for the majority of mutations in NPHP, screening in appropriate in children with typical clinical features. NPHP2, NPHP3, and NEK8 screening should be considered if age of presentation is less than 5 years. If a known genetic mutation is identified there is no need for a renal biopsy but should be considered if genetic screening is not available or if mutation is not identified. A diagnostic algorithm for genetic screening in NPHP is presented in figure 1.
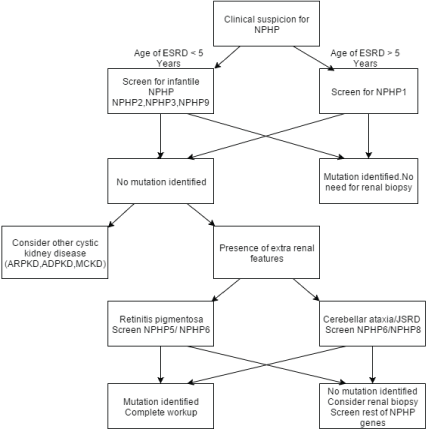
Figure 1: Diagnostic algorithm for genetic screening of NPHP [82]
ESRD – End Stage Renal disease NPHP – Nephronophthisis, ARPKD - Autosomal Recessive Polycystic Kidney Disease, ADPKD – Autosomal Dominant Polycystic Kidney Disease, MCKD – Multi Cystic dysplastic Kidney
Renal biopsy shows severe tubular damage. Tubular basement membrane abnormalities with varying degrees of tubular atrophy and interstitial fibrosis were seen [78]. Very few inflammatory cells are seen and glomeruli are usually normal in the early stages but will go on to have secondary sclerosis with progression of the disease.
Treatment
At present there is no definitive cure for NPHP and other related ciliopathies. Management centers on slowing progression of chronic kidney disease with optimal management of fluid balance, hypertension, proteinuria, metabolic bone disease, renal anemia, growth failure and timely renal replacement therapy. But with better understanding of ciliopathies and ongoing trial in animal models we could expect some definitive therapy in slowing renal cyst formation and progression in the future. Drugs of interest include but not limited to vasopressin receptor antagonist, mTOR inhibitors and cyclin dependent kinase inhibitors [79- 82] which have been tried with success in animal models of NPHP and ADPKD.
Conclusion
Our understanding of the molecular basis of NPHP has improved tremendously over the past decade. The role of primary cilia, cystoproteins in the pathogenesis and the fact that ciliopathies have a wide spectrum of clinical presentation has been learnt. The challenge still remains to understand the biological function of nephrocystins and the molecular mechanism behind cyst formation. Further research and understanding of the biology of cyst formation at a cellular level is necessary to facilitate the development of novel therapy to delay or reverse the disease process.
Conflict of Interest
The authors declare that they have no conflict of interest.
