Abstract
We present a case of a tumour in a 12-year-old mentally normal girl, who presented with a painless forehead swelling. Imaging studies demonstrated a large lesion within the right frontal lobe with erosion through the frontal bone. Histological diagnosis met all the criteria of glioblastoma with evidence of florid bone invasion. DNA methylation analysis, however, demonstrated features consistent with anaplastic pleomorphic xanthoastrocytoma. This skull invasion is a very uncommon presentation of gliomas, especially in children, and only a handful of cases have previously been described.
Keywords:
CNS tumors; Neuro-pathology; Tumors; Brain; Cancer genetics; Surgery; Neuro-oncology
Abbreviations:
MRI: Magnetic Resonance Imaging; GFAP: Glial Fibrillary Acidic Protein; S100: S100 protein; AE1/3: Cytokeratin AE1/3
stain; DNA: Deoxyribonucleic Acid; MIB-1: E3 ubiquitin protein ligase 1; EMA: Epithelial Membrane Antigen; CDKN2A: Cyclin-dependant kinase
inhibitor 2A; BRAF V600E: B-RAF proto-oncogene; ACNS: Trial number; WHO: World Health Organisation
Introduction
Pleomorphic xanthoastrocytoma is a rare World Health Organization (WHO) grade II tumour typically found in the temporal lobes in children and young adults. It has rarely been described with anaplastic features [1]. We present the first report of a previously healthy 12-year-old girl initially diagnosed histologically with frontal Glioblastoma with florid bone invasion through the skull. DNA methylation analysis subsequently showed features more consistent with anaplastic pleomorphic xanthoastrocytoma.
Case Report
The patient presented with a palpable, painless, non-pulsatile mass in the right supra-orbital region, which had developed over a 2-week period. She did not complain of headache and parents did not report any change in behaviour. Magnetic Resonance Imaging (MRI) scan demonstrated an ill-defined, avidly enhancing, and mass of heterogeneous signal intensity in the right frontal lobe causing 15 mm of midline shift (Figure 1). There was destruction of adjacent calvarium with extension into the sub-galeal soft tissue along with evidence of breech of the orbital roof with extension of the mass inferiorly into the orbit. MRI spine was unremarkable. At surgery, a grey-brown necrotic mass was seen protruding from the frontal bone. The orbital roof breech was explored, necrotic-appearing bone was resected and the abnormal tissue removed from the orbit. The remainder of the tumour was debulked and the orbital roof reconstructed. The frontal bone was replaced after excising a 1 cm margin at the defect. The patient awoke with no obvious deficits and an MRI day 1 post-operatively demonstrated no complications with a small enhancing area at the sylvian fissure (Figure 1).

Figure 1: (A) Pre-operative axial T1-weighted post contrast MRI demonstrating a right frontal heterogenous lesion with mass effect and erosion through the frontal bone. (B) Post-operative axial T1-weighted post contrast MRI demonstrating tumour resection and small area of enhancement posteriorly
Histopathologic examination revealed a tumour with a range of phenotypes, the predominant component being sheets of cells with oval to spindled nuclei and a coarse chromatin pattern but no obvious nucleoli. These cells showed moderate amounts of eosinophilic cytoplasm and mitotic figures were readily identified. Appearances were suggestive of meningothelial differentiation, however there were no clearly defined whorls, the nuclei lacked pseudo-inclusions and there was evidence of endothelial proliferation and florid pseudo-palisading necrosis (Figure 2A). The tumour showed extensive leptomeningeal spread and invasion into bone (Figure 2B). Stains for glial fibrillary acidic protein (GFAP), S100 protein, vimentin and, focally, for synaptophysin were positive. The E3 ubiquitin protein ligase (MIB-1) showed a proliferative index of 9%. AE1/3 immunostain highlighted a focal collection of rhabdoid cells. The epithelial membrane antigen (EMA) immunostain was negative, as were the immunostains for cytokeratin marker (CAM 5.2), cadherin, progesterone receptor and myogenin. Chromosome analysis showed gains of chromosomes 5,7,9,11,14,17&21. Loss of heterozygosity was not detected at 1p19q. While histologic diagnosis met all the criteria of Glioblastoma, the unusual finding of florid bone invasion, prompted further analysis using DNA methylationprofiling using the Illumina 450 k array (Illumina Human Methylation 450 k Array and Internal Classifier V7.0.). This revealed focal homozygous deletion of chromosome 9p, including the locus for cyclin-dependant kinase inhibitor 2A (CDKN2A) and classified the tumor as a Pleomorphic Xanthoastrocytoma. Sanger sequencing detected a proto-oncogene (BRAF V600E) point mutation. Integrating the histological features of a Glioblastoma, the florid bone invasion, the presence of a BRAF mutation, and the DNA methylation profile, a diagnosis of pleomorphic xanthoastrocytoma with anaplastic features was made.
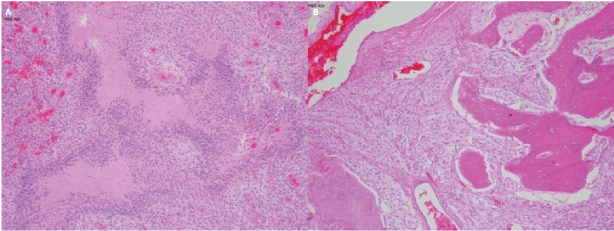
Figure 2: (A) H&E stain demonstrating endothelial proliferation and florid pseudo-palisading necrosis. (B) H&E stain demonstrating extensive leptomeningeal spread and invasion into bone.
The patient was treated with focal radiotherapy (59.4 Gy) with concomitant temozolomide as per the Children’s Oncology Group (COG) ACNS 0423 Protocol [2]. Towards the end of the radiotherapy she developed deep bone marrow aplasia which prevented the planned use of maintenance therapy with temozolomide and lomustine. After a protracted period, her marrow recovered. The patient remains clinically stable with evidence of stable minimal residual lesions in the tumour debulking site, more than two years after diagnosis. Given the promising results of BRAF inhibitors for central nervous system tumours positive for BRAFV600E mutations, these would be used should thetumour progress [3-6].
Discussion
A WHO grade for this rare entity is not clearly assigned [7-9]. Whilst the presence of a fibrous component raises the possibility of a gliosarcoma or of a desmoplastic infantile astrocytoma (DIA), this area was not obviously malignant and was not immunopositive for GFAP. Dura was involved macroscopically in this case however it has also been described to be macroscopically normal in cases with clear bony erosion [10]. This is thought to be due to microscopic invasion by glial cells. Low grade astrocytomas, oligodendrogliomas, and dysembryoblastic neuroepitheal tumours are known to cause bony remodelling or erosion of the inner table of the skull due to their slow growth. In contrast, calvarial invasion has been rarely described in malignant gliomas, having been reported in only a few case reports restricted to adult patients [10-12] and has never been reported in children with pleomorphic xanthoastrocytoma with anaplastic features.
Conclusion
The diagnosis of high grade glioma should be considered in cases where bony erosion and subgaleal extension is seen on imaging, especially if the tumour appears intra-axial. DNA methylation analysis had a significant clinical impact by enabling the accurate diagnosis of a very rare tumour entity. Our case highlights the need to establish a classification system which combines clinical, histopathological characteristics and also incorporates molecular analysis, such as DNA methylation profiling, to more accurately diagnose tumor entities.
Conflict of Interest Statement
The authors declare that they have no conflict of interest in regards to this case.
Download Provisional PDF Here
Article Information
Article Type: Case Report
Citation: Jukes A, Molloy C, Santoreneos C, Manton N, Koszyca B, et al. (2016) Brief Report: Anaplastic Pleomorphic Xanthoastrocytoma Invading the Skull in a Child. J Neurol Neurobiol 2(4): doi http://dx.doi.org/10.16966/2379-7150.128
Copyright: © 2016 Jukes A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 24 Aug 2016
Accepted date: 30 Sep 2016
Published date: 06 Oct 2016