Introduction
Improving immunogenicity and subsequent efficacy of vaccines is an on going challenge. For a successful induction of cell-mediated immune responses, pathogen-derived peptides should be efficiently generated and delivered to the MHC class I- and class II-restricted pathways [1,2]. Due to the differences in subcellular localization of peptide loading between MHC class I and class II, targeting immunogens to the cytoplasm and into the ER may result in an enhancement of CD8+ and CD4+ T-cell responses, respectively. In addition, directing pathogen-derived proteins for secretion may improve antibody induction and cross-presentation for elicitation of T cells [3,4]. Therefore, deliberate manipulation of the immunogen intracellular trafficking with the aim to improve vaccine performance is an area of active investigation.
The tissue plasminogen activator signal peptide (tPA-SP) is one of the most commonly used heterologous leader sequences for increasing expression levels of recombinant proteins in mammalian cells [5-12]. When coupled to the protein N-termini, it directed nascent growing amino acid chains into the ER and the cellular secretory pathway [5,13- 15]. Furthermore, modification of the tPA-SP cleavage signal facilitated its efficient removal and accelerated carrier nascent polypeptide folding into functional protein [16]. Although not beneficial for all coupled proteins [7,14], fusion of the tPA-SP with vaccine immunogens augmented induction of both antibody [5,9,15,17] and cell-mediated [9,11,18] responses, prolonged immunological memory [5,15] and conferred protection against pathogens in model infections [5,8,9,15,19].
We are developing a candidate vaccine against HIV-1, which focuses CD8+ T-cell responses on highly conserved regions of the HIV-1 proteins [20]. These are shared among most HIV-1 variants, mutations in them are frequently associated with replicative cost [21-24], but contain typically subdominant epitopes in natural infection. Following preclinical studies [20,25-34], vaccines delivering the HIVconsv immunogen have entered eight clinical trials in HIV-1-negative and positive European and African adults and induced frequencies of conserved region-specific T cells reaching median peaks between of 3-5.5 thousands of cells per million of circulating PBMCs for the most immunogenic regimens [35-38]. In these studies, the gene coding for the HIVconsv immunogen was vectored by plasmid DNA, non-replicating simian (chimpanzee) adenovirus ChAdV-63 and non-replicating poxvirus modified vaccinia virus Ankara (MVA) and had no leader peptide. Because the frequencies of effective T cells necessary to achieve protection of humans against HIV-1 are not known [39,40], our ongoing aim is to improve the vaccine performance. In this study and to inform future development, we investigated whether or not the T-cell induction by the HIVconsv vaccines benefits from attachment of the tPA-SP to the HIVconsv immunogen.
Materials and Methods
Construction and preparation of vaccines
Design, construction and preparation of the HIVconsv [20] and tHIVconsv [25] vaccines were described previously. For immunization, the plasmid DNA was produced in E. coli DH5α, isolated using EndoFree Plasmid Giga kit (Qiagen) and stored at -20°C until use. Recombinant ChAdV-63 vaccines were grown in suspension culture of HEK 293 cells, purified by chromatography, titred and stored at -80°C until use. Recombinant MVA vaccines were grown in primary chicken embryo fibroblasts, purified by two CsCl cushion centrifugations, titred and stored at -80°C until use.
Immunofluorescent staining
HEK 293T cells grown on coverslips in a 6-well plate were transfected with 2.5 µg of pSG2.tHIVconsv or pSG2.HIVconsv plasmid DNA using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s specifications. For infections, HEK 293T cells were infected with recombinant MVA or ChAdV63 expressing HIVconsv or tHIVconsv proteins at a multiplicity of infection (MOI) of 5 and 10, respectively, in serum free DMEM medium at 37°C, 5% CO2 for 2 hours. Cells were then washed with PBS, incubated in complete DMEM medium (10% FCS and 1% Penicillin/Streptomycin) for further 24 hours, washed with ice cold PBS and fixed for 10 min. on ice with 10% neutral buffered formalin solution containing 4% formaldehyde (Sigma). Fixed cells were incubated at room temperature for 20 min., washed with PBS and permeabilized with 0.2% Triton (TX-100) for 5 min., washed again, blocked with 1% bovine serum albumin (BSA) in PBS for 30 min. with anti-Pk (Abcam), anti-calnexin (BD Bioscience) and anti-CD63 mAb (BD Bioscience) mAbs. The cells were washed 3x for 15 min., mounted on microscope slides with the Vectashield DAPI nuclear stain mounting media (Vector Laboratories) and then examined on a fluorescence microscope (Leica DMI 3000B).
Pulse-chase analysis
Metabolic labeling of DNA-transfected cells with [35S]methionine was described previously [41]. Briefly, HEK 293T cells were transiently transfected with DNA as described above. After 18 hours, growth medium was replaced with methionine-free minimum essential medium for 30 min. prior to the addition of fresh medium containing 75 µCi (2.7 MBq) [35S]methionine ml-1. After a 15 min.-labeling period, the cells were incubated further for various times in medium containing unlabeled methionine and lysed in a lysis buffer containing Nonidet P-40, 100 mM iodoacetamide and protease inhibitors.
Immunoprecipitation and SDS-PAGE
Cell lysates were analyzed by immunoprecipitation followed by SDS-PAGE, as described previously [41]. Briefly, cell lysates were immunoprecipitated with an anti-Pk-tag mAb (Abcam) and protein A– Sepharose overnight at 4°C. Cells were pelleted and pellets were washed, resuspended in SDS sample buffer and heated at 95°C for 5 min. prior to SDS-PAGE on a 10% acrylamide reducing gel.
Mice, immunizations and preparation of splenocytes
Groups of 5- to 6-week-old female BALB/c mice were used. All vaccine administrations were carried out intramuscularly under general anaesthesia with 100 µg of DNA, 106 plaque-forming units (PFU) of recombinant MVA and 3 × 108 infectious units (IU) recombinant ChAdV63. On the day of sacrifice, spleens were collected and cells isolated by pressing organs individually through a nylon cell strainer (BD Falcon) using a 5-ml syringe rubber plunger. Following the removal of red blood cells with RBC Lysing Buffer Hybri-Max (Sigma), splenocytes were washed and re-suspended in R10 (RPMI 1640 supplemented with 10% FCS, penicillin/streptomycin and β-mercaptoethanol) for ELISPOT, intracellular cytokine staining (ICS) assays and other procedures.
Ethics statement
All animal procedures and care conformed strictly to the United Kingdom Home Office Guidelines under The Animals (Scientific Procedures) Act 1986. The protocol was approved by the local Research Ethics Committee (Clinical Medicine, University of Oxford). Experiments were carried out under Project Licence no. 30/2833 held by TH with a strict implementation of the Replacement, Reduction and Refinement (3Rs) principles.
Peptides and peptide pools
All peptides were at least 80% pure by mass spectrometry (Ana Spec, San Jose, CA, USA), were dissolved in DMSO (Sigma-Aldrich) to yield a stock of 10 mg/ml, and stored at –80°C. One hundred and ninety nine HIVconsv-derived peptides (15/11) were divided into 6 pools of 32 to 35 individual peptides for use in ICS and ELISPOT assays. The peptides were used at a final concentration of 1.5 µg/ml.
IFN-γ ELISPOT assay
The ELISPOT assay was performed using the Mouse IFN-γ ELISpot kit (Mabtech) according to the manufacturer’s instructions. Spots were visualised using sequential applications of a biotin-conjugated secondary anti-IFN-γ mAb (R4-6A2, Rat IgG1), an alkaline phosphatase and a chromogenic substrate (Bio-Rad), and counted using the AID ELISpot Reader System (Autoimmun Diagnostika). Background no-peptide frequencies of IFN-γ-producing cells were below 40 SFU/106 splenocytes and were subtracted from those of peptide-stimulated cultures.
Intracellular Cytokine Staining (ICS)
One million cells were stimulated with peptides or peptide pools at 37°C, 5% CO2 for 90 min., before adding GolgiStop (BD bioscience). If required, CD107a-FITC antibody was added at the start of stimulation. After 5-hr incubation, the cells were washed with FACS buffer (PBS, 1% FCS, 0.01% Azide) and blocked with anti-CD16/32 antibodies (eBioscience) at 4°C for 20 min., then stained with anti-CD8 and anti-CD4 mAb (eBioscience). The cells were then washed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) and stained for intracellular cytokines: anti-TNF-α, anti-IFN-γ and anti-IL-2 mAb (eBioscience). The cells were washed and fixed with Cell Fix (BD Biosciences), then acquired using LSRII (BD) flow cytometer and analysed with Flowjo (Tree Star) and SPICE software.
Ex-vivo killing assay
Equal numbers of P815 cells were differentially labelled with either 800 nM or 32 nM of CFSE according to the manufacturer’s specifications. The P815 cells labelled with 800 nM CFSE were pulsed with peptides for 2 hours and washed several times. Splenocytes from immunized mice were prepared as described above, mixed with the differentially CFSElabeled P815 target cells at an effector-to-target (E:T) ratio of 10:1 or 5:1 and incubated overnight at 37°C. The cells were washed, stained with a live/dead marker and analyzed using flow cytometry. Cytotoxicity was calculated using the following formula: % specific lysis=100 × (number of unpulsed control cells–number of peptide-pulsed cells)/number of unpulsed control cells.
Statistical analysis
Statistical analyses were performed using Graph Pad Prism version 5. T-cell responses were assumed to be non-Gaussian in distribution, thus results are presented as medians (range). Multiple comparisons were performed using the Kruskal-Wallis test. Groups with the same in vitro restimulations were compared using two-tailed Mann-Whitney U tests. A P value<0.05 was considered significant.
Results
tPA-SP fusion to HIVconsv increases protein expression, but
not half-life, after DNA transfection
The 806-amino acid-long sequence of the HIVconsv immunogen is published [20]. DNA sequence coding for the 22P/A-mutated signal sequence of tPA (MDAMKRGLCCVLLLCGAVFVSAR) [16] was attached to the 5’-end of the HIVconsv open-reading frame (ORF) to express tHIVconsv (Figure 1A). Particularly the 22P/A-mutated tPA-SP will be efficiently co-translationally removed during translocation into the ER by signal peptidases [25,42]. For the DNA vaccine modality, we first used microscopy to assess the subcellular localization of the HIVconsv protein, or more precisely its C-terminal Pk tag [43], in transiently transfected HEK 293Tcells and did so relative to calnexin, an ER chaperon protein, and CD63, a late endosome/lysosome marker. The immunofluorescence analysis showed a brighter staining for tHIVconsv-transfected cells relative to those expressing the unmodified HIVconsv, and suggested a co-localization of the Pk tag with calnexinin ER enhanced by the tPASP without reaching the late endosomal/lysosomal compartments (Figure 1B). Flow cytometry analysis identified a higher frequency of HEK 293T cells positive for the Pk tag after transfection with the pSG2.tHIVconsv relative to the pSG2.HIVconsv vaccines and showed a trend in the same direction for higher mean fluorescent intensity, again suggesting higher expression of the tPA-SP-modified HIVconsv (Figure 1C). The intracellular half-life of the undegraded HIVconsv protein was found to be similar following the pSG2.HIVconsv and pSG2.tHIVconsv transfections in a pulse-chase experiment with slightly increased protein levels at 0, 0.5 and 1 hour for the latter (Figure 1D). Collectively, these data suggested that following DNA delivery, addition of the tPA-SP to HIVconsv allowed for a higher HIVconsv protein accumulation in the ER of the transfected HEK 293T cells.
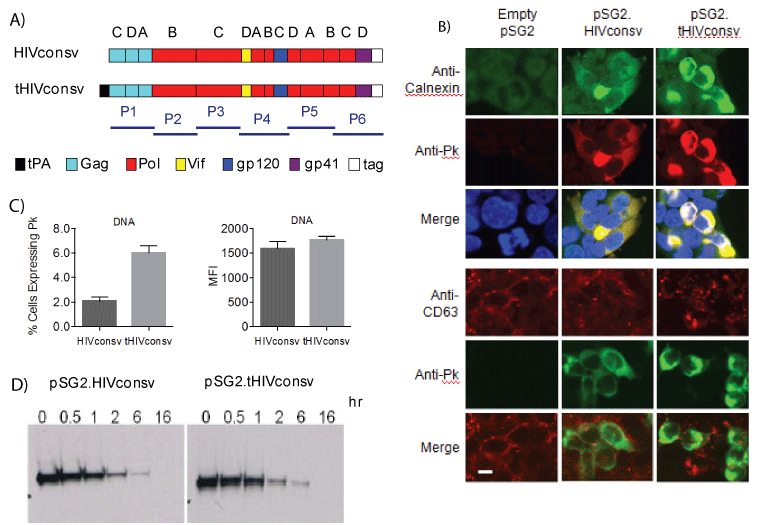
Figure 1: Expression of the HIVconsv immunogen from the pSG2. HIVconsv and pSG2.tHIVconsv DNA vaccines. (A) Schematic diagram of the HIVconsv and tPA-SP-fused tHIVconsv immunogens. T-cell epitope H and mAb Pk tag were attached to the C-terminus of HIVconsv to facilitate pre-clinical development [20]. Letters above the conserved regions indicate the clade, the consensus of which was used, and bars bellow depict the areas of immunogens covered by peptide pools P1-P6. (B) Subcellular localization of the HIVconsv protein (Pk tag). HEK 293T cells were transfected with the pSG2.HIVconsv and pSG2.tHIVconsv vaccine DNAs. Two days later, cells were incubated with chromogenconjugated mAbs specific for the Pk tag (red on the top and green on the bottom panel), calnexinas an ER marker (green) and CD63 as a late endosomal/lysosomal marker (red) to characterize the subcellular localization of HIVconsv immunogen. DAPI was used to visualize the nuclei (blue). Cells were photographed using laser scanning confocal microscopy.Bar, 20 µm. (C) Two days after DNA transfection, HEK 293T cells were lifted from the plates and incubated with FITC-conjugated anti-Pk mAb to detect HIVconsv expression. Percentages of HIVconsvexpressing cells (left) and their MFI (right) were determined using flow cytometry (see Figure 3 for a representative gating). Data are shown as mean ± SEM (n=3). (D) Analysis of the HIVconsv protein half-life in HEK 293T cells transfected with either pSG2.HIVconsv or pSG2. tHIVconsv DNA vaccines. After metabolic labeling with [35S]methionine, cells were chased with cold methionine for the indicated times above the autoradiographs and cell lysates were analyzed by immunoprecipitation using anti-Pk mAb followed by 10% SDS-PAGE and autoradiography.
tPA-SP-modification of HIVconsv improved T-cell induction by
DNA vaccines
To investigate the effect of tPA-SP on T-cell immunogenicity, groups of BALB/c mice were vaccinated with 1, 2 or 3 doses of recombinant DNA (D, DD or DDD regimens) and the induction of CD8+ T cells was analyzed by intracellular IFN-γ staining initially using H-2d immunodominant peptide H (RGPGRAFVTI; Env residues 311-320) [44] added for the purpose of preclinical development to the C-terminus of HIVconsv [20] (Figure 1A). Immunization with the pSG2.tHIVconsv DNA induced 2- to 3.5-fold higher frequencies of H-specific CD8+ T cells relative to the leaderless pSG2.HIVconsv vaccine, which reached a statistical significance for the D (P=0.01) and DD (P=0.02) regimens (Figure 2A). In separate immunizations, we assessed the breadth and specificity of elicited T cells. Mice received the DDD regimen and for each of the pSG2.tHIVconsv and pSG2.HIVconsv groups, cells were enumerated by responding to six pools P1-P6 of 32-35 overlapping 15-mer peptides spanning the entire HIVconsv protein in an IFN-γ ELISPOT assay [45]. Responses were detected to pools P2, P4 and P6 ranging from 448-1055 and 128-415 spot-forming units (SFU)/106 splenocytes for the pSG2. tHIVconsv and pSG2.HIVconsv vaccines, respectively, with significantly higher frequencies induced by the modified tHIVconsv (Figure 2B). Also 75 SFU/106 splenocytes specific to P1 were induced by the tHIVconsv, but not by the HIVconsv delivery. Analysis of H-specific T cells in ICS for production of IFN-γ, TNF-α and IL-2 production and degranulation (CD107a) detected plurifunctionality within both populations and the overall tPA-SP-enhancement of immunogenicity was consistent with the IFN-γ ELISPOT assay (Figure 2C). In a separate DDD vaccination, this time using pSG2.HIVconsv, pSG2.tHIVconsv and pSG2.HIVconsv+pSG2. HIVconsv (half-doses), immune splenocytes were tested against all 199 individual peptides across HIVconsv, reasoning that different subcellular localization of the HIVconsv protein may lead to differential epitope processing and presentation, and using the unmodified and tPA-SPcoupled immunogens together may benefit the overall breadth of response. Thus, the strongest responses among clusters of adjacent overlapping 15-mers were detected to peptides 15, 42, 112 and 197 (containing the H epitope). There was a hint of differential processing with HIVconsv inducing stronger responses to peptide 112 compared to 42, while the tHIVconsv immunogen elicited higher frequencies of T cells to peptide 42 relative to 112, while mixed HIVconsv+tHIVconsv delivery induced equal peptide 42 and 112 frequencies. However, Kruskal-Wallis test did not find the three sets of T-cell frequencies induced by different immunogens and their combination significantly different (P=0.19) (Figure 2D).

Figure 2: tPA-SP enhances HIVconsv immunogenicity for plasmid DNA delivery. (A) Groups of BALB/c mice were immunized with either pSG2. HIVconsv (dark) or pSG2.tHIVconsv (light) DNA once (D), twice (DD) or three times (DDD) in 7-week intervals and sacrificed 2 weeks after the last immunization. CD8+ T cell induction was assessed in an ICS assay using the optimum H-2d immunodominant peptide H. (B) In a separate experiment, mice received 3 DNA immunizations (DDD) using either the pSG2.HIVconsv (dark) or pSG2.tHIVconsv (light) vaccines. Their HIVconsv-specific T cells were enumerated in an IFN-γ ELISPOT assay employing six peptide pools P1-P6 of 15-mer peptides overlapping by 11 amino acids spanning the entire HIVconsv protein (Figure 1A). (C) Immune splenocytes were analysed using polychromatic flow cytometry for production of IFN-γ, TNF-α, IL-2 and degranulation (CD107a) upon restimulation with epitope H. Percentages of H-specific T cells induced by vaccines expressing HIVconsv (dark) and tHIVconsv (light) are indicated on the left and the T-cell plurifunctionality is shown on the right. See Figure 4E for a representative gating strategy. Data in (A-C) are shown after subtracting no-peptide background as median (interquartile range) (n=5). For the same in vitro restimulations, the HIVconsv and tHIVconsvinduced frequencies of H- or pool-specific T cells were compared using a two-tailed Mann-Whitney U test, whereby asterisk indicates P<0.05. (D) Groups of BALB/c mice received 3 doses of either pSG2.HIVconsv (top), pSG2.tHIVconsv (middle) or half doses of mixed pSG2.HIVconsv+pSG2. tHIVconsv (bottom) as indicated above the graphs and their pooled HIVconsv-specific splenocytes were enumerated against199 individual 15-mers peptides (only peptides with spots in the wells are shown) (n=3). Dotted line indicates the arbitrary threshold for a positive response (at least 50 SFU/106 above the background). Kruskal-Wallis test was used to comparethe three sets of T-cell frequencies induced by the unmodified and tPA-SP-coupled immunogens and their combination (P=0.19).
Distinct patterns of HIVconsv expression with and without tPASP expressed from viral vectors
The core of our clinical strategy for delivery of T-cell immunogens consists of simian adenovirus prime and MVA boost. Therefore, genes coding for the HIVconsv and tHIVconsv immunogens were engineered into these two vectors and the vaccines were designated ChAdV63. HIVconsv, ChAdV63.tHIVconsv, MVA.HIVconsv and MVA.tHIVconsv. Similar to the DNA vaccines, the Pk tag expression was confirmed by immunofluorescence using the Pk-specific mAb. Here when using viral vectors, an accumulation of fluorescence in cytoplasmic vesicular structures was more pronounced for the tPA-SP vaccines (Figure 3A). In flow cytometry, the effect of tPA-SP on HIVconsv expression differed in recombinant ChAdV63- and MVA-infected cells. While moderately higher numbers of cells infected with MVA.HIVconsv than MVAtHIVconsv expressed the Pk tag and did so with slightly higher MFI, these parameters were inverted back in favour of the tHIVconsv delivery for the recombinant ChAdV63 infection (Figure 3B). Multiple time points and multiplicities of infection were tested for both viruses and 24 hours post-infection was selected as an optimal compromise between expression levels and cells loosing adherence due to cytopathic effect (not shown). Thus at least at this time point, the effect of tPA-SP addition on the HIVconsv expression differed for the poxvirus and adenovirus vectors.
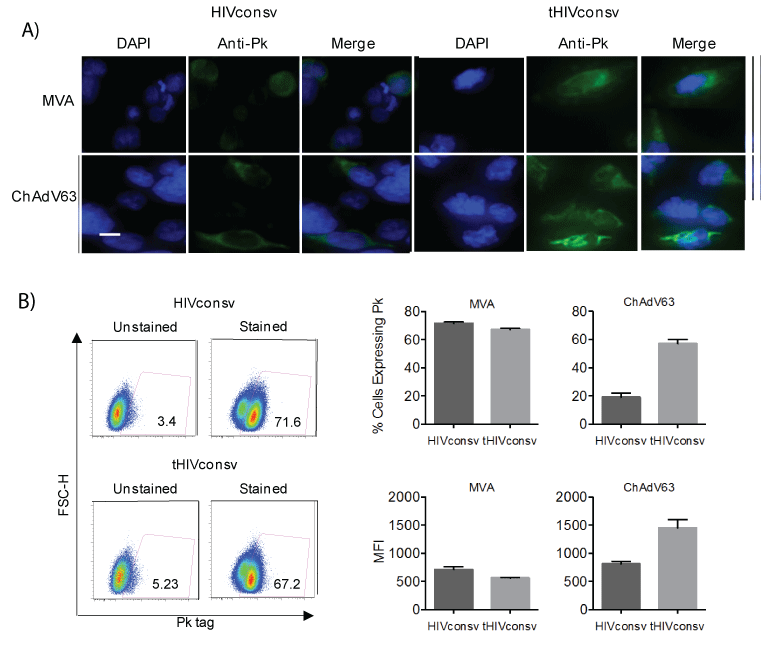
Figure 3: Expression of HIVconsv without or with tPA-SP from the MVA and ChAdV-63 vaccines. (A) Subcellular localization of HIVconsv expressed unmodified and with tPA-SP in HEK 293T cells infected with poxvirus-(MOI 5) or adenovirus-(MOI 10) vectored vaccines for 24 hours was visualized using FITC-conjugated anti-Pk tag mAb (green) and confocal microscopy. DAPI localized the cell nuclei (blue). Bar, 20 µm. (B) One day after infection, HEK 293T cells were lifted and incubated with FITC-conjugated anti-Pk mAb, and using the depicted gating (left), the percentages of HIVconsv-expressing cells and their MFI (right) were determined using flow cytometry. Data are shown as mean ± SEM (n=3).
Weak negative effect of tPA-SP on T-cell induction by the ChAdV63-MVA regimen (CM)
To assess the effect of the tPA-SP on induction of HIVconsv-specific T cells by the ChAdV63-MVA regimen, groups of BALB/c mice were immunized sequentially with the ChAdV63 and MVA vaccines coding either for the HIVconsv or tHIVconsv immunogen (Figure 4A). The IFN-γ ELISPOT assay employing HIVconsv peptide pools P1-P6 detected a less that 2-fold decrease in T-cell frequencies by addition of the tPASP. Decrease in the response magnitude was only significant for pools P2 (P<0.01) and P5 (P<0.03) (Figure 4B). Peptide-stimulated T-cell frequencies showed overall similar trend, which reached significance for peptides 42 (P=0.007) and 112 (P<0.001) (Figure 4C). For the three most immunodominant peptides 42, 112 and H, ex vitro killing assays were performed to determine if the tPA-SP improves CD8+ T-cell lytic activity against peptide-pulsed target cells. While strong lysis at effector to target ratio 10:1 was detected with median killing of between 44% and 77%, statistically stronger killing was only detected for peptide H and this was in favour of the unmodified HIVconsv immunogen (Figure 4D). Next, we assessed whether or not the addition of tPA-SP to the HIVconsv immunogen in the CM regimen affected the expression of IFN-γ, TNF-α and IL-2 by CD8+ T cells. While an approximately 2-fold significant decrease was detected for peptide 112-specific CD8+ T cells producing IFN-γ, TNF-α and IL-2 after tPA-SP attachment, a significantly difference for peptide H was only detected for IL-2 production and no difference for peptide 42 was found for any function (Figure 4E). Thus, for the CM regimen, tPA-SP addition to HIVconsv weakly decreased T-cell induction for some epitopes.
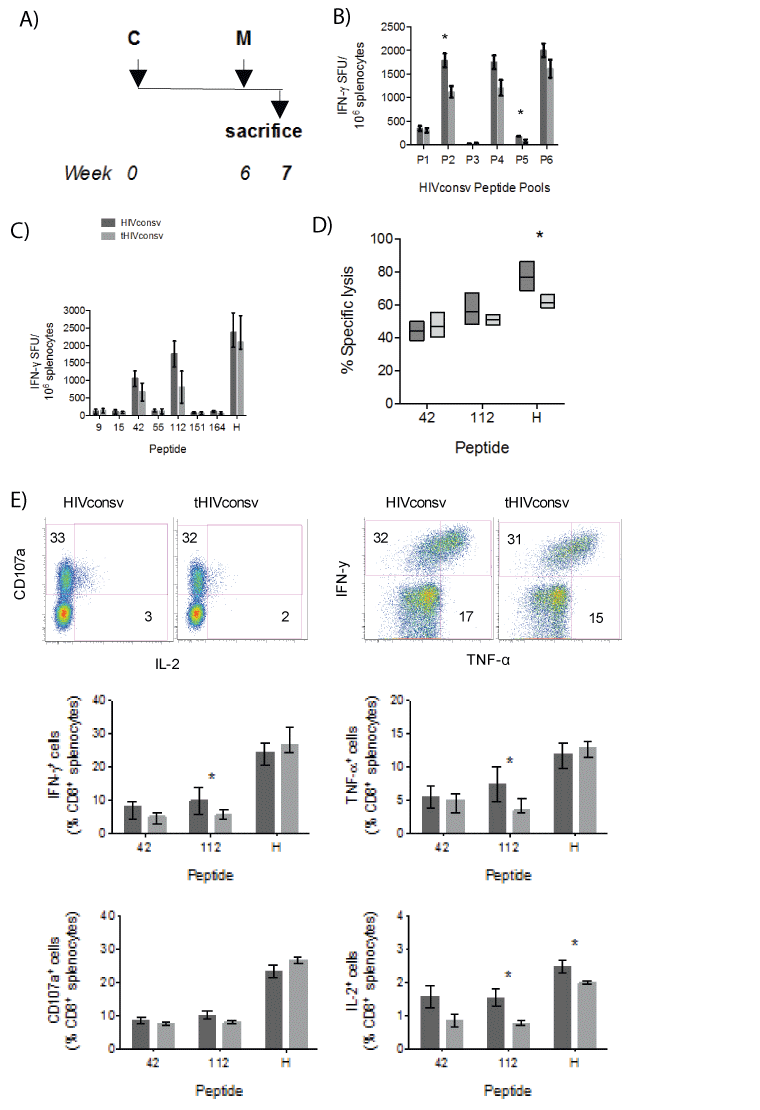
Figure 4: A weak negative effect of the tPA-SP addition on HIVconsv immunogenicity in the ChAdV-MVA (CM) regimen. (A) The immunization schedule. (B) Groups of BALB/c mice were immunized with recombinant ChAdV63 and MVA vaccines expressing either the unmodified (dark) or tPA-SP-linked (light) HIVconsv immunogen. Effect of the tPA-SP on the HIVconsv immunogenicity was assessed in an IFN-γ ELISPOT assay using six peptide pools P1-P6 (B) and selected individual stimulatory 15-mer peptides and peptide H (C) (n=5+5; pooled from 2 experiments). (D) The same splenocytes were assessed for their ability to lyse P815 target cells pulsed with the two most immunodominant 15-mer and the H peptides at effector-to-target ratio 10:1 (n=5). (E) Immune CD8+ T cells were analysed using polychromatic flow cytometry for production of IFN-γ, TNF-α, IL-2 and degranulation (CD107a) upon restimulation with peptides 42, 112 and H (n=5). For all graphs, data are shown after subtracting background as median (interquartile range). The HIVconsv and tHIVconsv-induced T-cell frequencies detected with the same pool/ peptide restimulation were compared using two-tailed Mann-Whitney U tests. Asterisks indicate P<0.05.
Minimal effect of tPA-SP on T-cell induction by the DNA-DNADNA-ChAdV63-MVA regimen (DDDCM)
Next, the effect of tPA-SP on T-cell immunogenicity was determined following three DNA primes boosts by recombinant vectors ChAdV63 and then MVA (regimen DDDCM; Figure 5A). Using an accelerated 3-week gap between DNA administrations and a standard 6-week interval after recombinant ChAdV63, significant differences in IFN-γ ELISPOT assay-determined T-cell frequencies between mice receiving tHIVconsv and those receiving HIVconsv were not observed for any of the six peptide pools (Figure 5B) nor for peptides tested individually (Figure 5C). A significantly stronger ex-vivo killing of peptide-pulsed P815 target cells by splenocytes isolated from mice, which had received tHIVconsv, was only detected for peptide H (P=0.029) (Figure 5D). Finally, direct comparison of the DDD, CM and DDDCM regimens was performed employingthe IFN-γ ELISPOT assay and more recently defined optimal peptides LVGPTPVNI (in 15-mer peptide 42) and (YYDPSKDLI (in 15-mer peptide 112) [31]. While the overall trend in frequencies of specific T cells induced by the DDD and DDDCM regimens of tHIVconsv>HIVconsv and the inverted trend of tHIVconsv and the inverted trend of tHIVconsv<HIVconsv for the CM regimen were
noticeable, only a few comparisons reached statistical significance with
less than 2-fold differences in median magnitudes (Figure 5E).
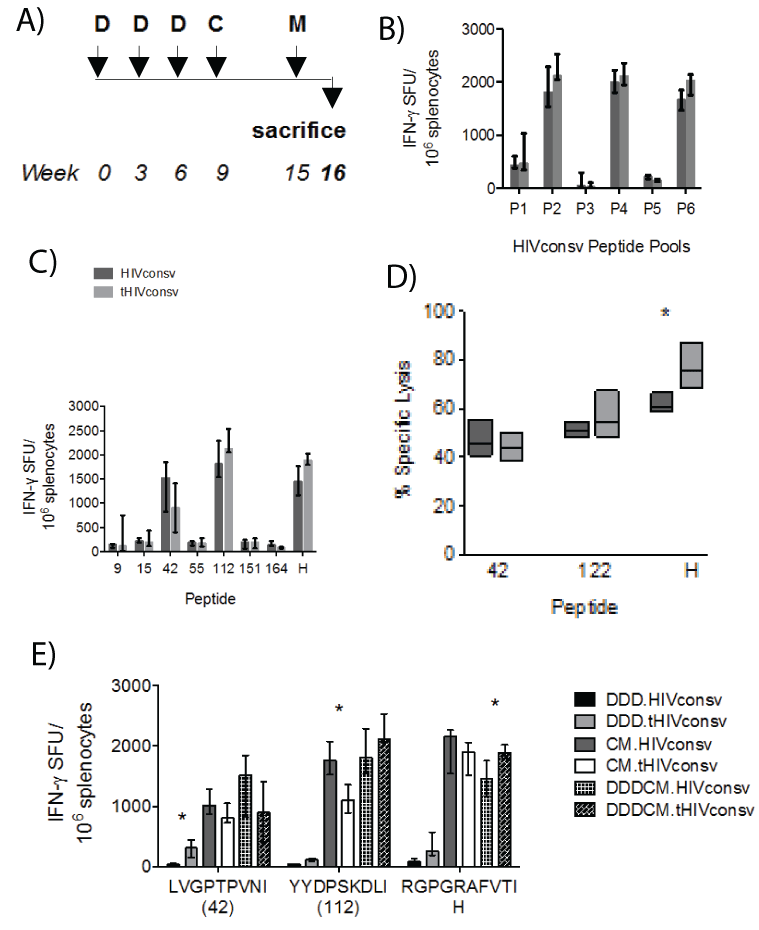
Figure 5: Lack of a consistent effect of the tPA-SP on T-cell induction by HIVconsv in the DNA-DNA-DNA-ChAdV-MVA (DDDCM) regimen. (A) The immunization schedule. (B) Groups of BALB/c mice received either the HIVconsv (dark) or tHIVconsv (light) immunogens delivered by the DDDCM regimen and the frequencies of HIVconsv-specific T cells were assessed in an IFN-γ ELISPOT assay using six peptide pools P1-P6 (B), and individual responding 15-mer peptides and peptide H (C) (n=4). (D) Splenocytes were tested for the ability to lyse P815 target cells pulsed with the two most immunodominant 15-mer peptides and peptide H at effector-to-target ratio 10:1 (n=5). (E) Mice received one of the DDD, CM or DDDCM regimens and the elicited frequencies of T cells specific for the three most immunogenic epitopes were assessed in an IFN-γ ELISPOT assay using the optimal-length peptides (n =3). For all graphs, data are shown after subtracting background as median (interquartile range). The HIVconsv and tHIVconsv-induced T-cell frequencies detected with the same pool/peptide restimulation were compared using two-tailed MannWhitney U tests. Asterisks indicate P<0.05.
Discussion and Conclusion
Since their construction [20], we have reported on the high T-cell immunogenicity of the conserved-region HIVconsv vaccines demonstrated in mouse, non-human primate and phase I/IIa human clinical studies, where their administration induced high-frequencies relative to other studies of broadly-directed, plurifunctional CD8+ T-cell responses [20,25,28,30-36,38]. Nonetheless, we continue searching for strategies capable of further improvement of the vaccine performance to maximize the chance for efficacy in humans. Here, we assessed in mice the effect on T-cell immunogenicity of directing the HIVconsv nascent polypeptide into the ER and the secretory pathway by addition of the tPA-SP. For all three tested vaccines pSG2.tHIVconsv DNA, ChAdV63. tHIVconsv and MVA.tHIVconsv, we detected augmented HIVconsv protein levels in transfected/infected humans cells and its higher accumulation in vesicular cytoplasmic structures. While this modification increased 2- to 3.5-fold the frequencies of T cells induced by the pSG2. tHIVconsv DNA, this benefit was lost for a much more potent delivery using the ChAdV63.HIVconsv-MVA.HIVconsv heterologous primeboost regimen. No induction of HIVconsv-specific antibodies was ever detected in immunized mice (not shown).
Our results are well in agreement with published studies. Immunogens fused to the tPA-SP showed increased protein expression and secretion with a concomitant enhancement of immune responses and, in some cases, increased protective efficacy [7-11,14,15,19,46-48]. All these positive reports have almost invariably two features in common: they employed vaccines vectored by plasmid DNA and the improved vaccine performance was observed in the mouse model with the exception of Casimiro et al. [49] who observed the benefit of tPA-SP addition in macaques, too. Thus, it remains open whether or not any vaccination benefits will transfer to humans. The different immunogenic properties of plasmid DNA as opposed to viral vectors were noted previously, whereby immunization with DNA and poxvirus led to difference in long-term epitope hierarchies [29]. Furthermore, a few other studies failed to detect an advantage of the tPA-SP on induction of immune responses [14,46,50]. Thus, the effect of tPA-SP addition is immunogen specific and will need to be tested and confirmed for each particular immunogen, vector modality, regimen and type of immune responses induced.
Although tPA-SP is a popularly used signal peptide for DNA vaccines, other cellular chaperones are under investigation for improvement of vaccine performance. Thus, more recently fusion of the MHC class IIassociated invariant chain(Ii) to various immunogens was reported to enhance CD8+ T-cell responses [51-53]. This enhancement was shown for vaccines vectored by adenovirus, could be translated from mice to nonhuman primates and vaccines delivering invariant chain-coupled hepatitis C virus-derived immunogen are currently in the pipeline for human trials. Also inclusion of the lysosome-targeting sequences of lysosomalassociated membrane protein-1 (LAMP-1) in DNA plasmid-vectored vaccines was shown to redirect immunogens from a proteasomal-MHC class I towards the lysosomal-class II pathway and led to a significant enhancement of the vaccine immunogenicity in several animal models [15,54,55]. Thus, the subcellular targeting strategy remains a highly active area of investigation for improving vaccine efficiency.
Overall, this study demonstrated that linking the tPA-SP to the HIVconsv immunogen increased its intracellular expression, but performance of the vaccines only benefited when the tHIVconsv was delivered by ‘naked’ DNA. For the already potent ChAdV63- and MVA-mediated delivery, the tPA-SP addition did not further increase the T–cell induction above that achieved by the unmodified HIVconsv. However, it is possible that targeting of the nascent HIVconsv protein into the ER/secretory pathway may translate into a lower toxicity to the vaccine-producing cells and, in turn, result in high vaccine yields during manufacture, although the latter remains to be demonstrated. While our conclusions concur with published observations for other DNA-vectored vaccines, the main novelty here is in the comprehensive comparison of three vaccine vector systems and establishment of the tPA-SP effect for the immunogenicity of our unique T-cell immunogen. Collectively, the available evidence from this and other studies suggests that the tPA-SP enhancement of vaccine immunogenicity cannot be assumed without confirming for particular immunogens, expression modalities, types of desired immune responses and intended species.
Acknowledgements
The work was jointly funded by the UK Medical Research Council (MRC G1001757 to T.H.) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreements. S.A.-J. was supported by the King Abdullah scholarship by the Ministry of Higher Education, Kingdom of Saudi Arabia. B.O. was funded in part by the International AIDS Vaccine Initiative (IAVI) and her funding was made possible by the support of the United States Agency for International Development (USAID) and other donors. The full list of IAVI donors is available at http://www.iavi.org. T.H. is the Jenner Institute Investigators. We are also grateful to IAVI for provision of the HIVconsv peptides.
The authors have no competing interests.
