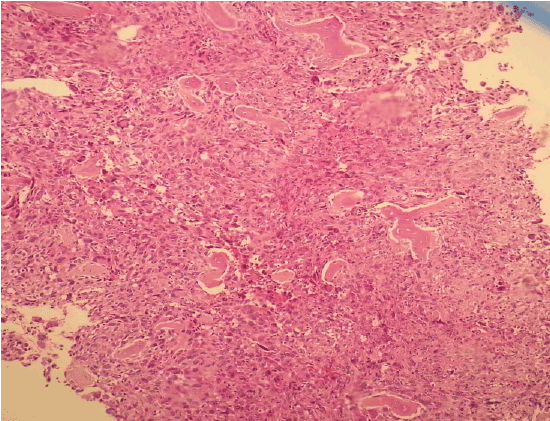
Figure 1: Young mast cell infiltration in bone marrow (H & E X100)
Elife Kimiloglu1* Zuhal Ozcan1 Nusret Erdogan2 Aysenur Akyildiz Igdem1 Osman Mavis3 Tayfun Elibol3
1Department of Pathology, Taksim Education and Research Hospital, Istanbul, Turkey*Corresponding author: Elife Kimiloglu, Department of Pathology, Taksim Education and Research Hospital, Istanbul, Turkey, E-mail: elife.sahan@gmail.com
Mastocytosis is a clonal disease derived from hematopoietic bone marrow progenitor cells that manifests with an unusually broad spectrum of clinical and morphological appearances.
We report the case of a 48-year old male patient who presented with hepatosplenomegaly for 12 years, yellow-brown colored maculopapular pigmentation on the skin and pancytopenia.
Materials of each finding were sent to us and we reported as diffuse mast cell infiltration for biopsy of bone marrow, portal mast cell infiltration for tru-cut biopsy of liver and urticaria pigmentosa (adult type) for punch biopsy of the skin. The diagnosis of systemic mastocytosis was confirmed by raised tryptase level.
Systemic mastocytosis; Hepatosplenomegaly
Mastocytosis is a rare and heterogeneous disease characterized by the presence of excessive numbers of mast cells in various organs, mainly the skin and the bone marrow that manifests with an unusually broad spectrum of clinical and morphological appearances [1,2].
Although bone marrow is the main tissue for diagnosis of systemic mastocytosis (SM), the demonstration of compact mast cell infiltrates in extramedullary tissues should be regarded as indicative of involvement by the disease [3,4].
To be able to separate the different forms of SM, pathologists must be aware of clinical symptoms, especially the so-called B findings, referring to an organomegaly (hepatosplenomegaly and/or lymphadenopathy), and even C findings, indicating organ dysfunction due to widespread mast cell infiltration (e.g., cytopenia and/or ascites in aggressive SM with strong infiltration of bone marrow and liver) [2] .
A 48-year old male patient who presented with hepatosplenomegaly for 12 years, yellow-brown colored maculopapular pigmentation on the skin (except his face) and pancytopenia. In biochemical analysis results increased levels of CRP (8,92 mg/L), sedimentation 60’(51 mm/h), GGT (64 U/L), LDH (224 U/L) and ASO (535,5 U/ml) were identified. The AST (SGOT), ALT (SGPT), lipase, amylase and 5-hydroxy indole acetic acid were within normal range.
In hemogram decreased levels of WBC (3,2-10^3/µl), RBC (3,87- 10^6/µl), Hemoglobin (10,3 g/dL), HCT (32,3%), Platelet (73-10^3/µl) and Lymphocyte (16,4%) were identified. Anti HBs, Anti HCV, Anti HIV, Coombs’ Wright test, Gruber-Widal agglutination, Brucella agglutination, VDRL RPR syphilis, TPHA syphilis and Monotest were negative.
Hepatomegaly-for 12 years (20 cm), splenomegaly (23,5 cm) and lymphadenopathy (Para-aortic, aorta caval, Para-celiac and portal) were evaluated on MRI and CT scanning and US of the abdomen. His mother had hepatomegaly that was not examined. The patient had yellow-brown colored maculopapular pigmentation on the skin except his face.
Bone marrow aspirate was empty. The sections of bone marrow biopsy stained with hematoxylin and eosin (H&E) showed diffuse solid infitration of young mast cells (Figures 1 and 2); staining with Giemsa (Figure 3) and Toluidine blue stained them meta chromatically (Figure 4). Immunohistochemistry was performed which showed positivity with CD117 and CD68 (Figure 5) in the mast cells; negativity with myeloperoxidase, CD34, S-100 and rare positivity with CD1a. CD3 and CD20 were positive on reactive T and B lymphocytes.
The histopathological studies of skin punch biopsy of the maculopapular lesions showed infiltration of the superficial dermis with mast cells (>40/40 High Power Field) staining with Giemsa and Toluidine blue (Figure 6) stained them meta chromatically therefore, diagnosed as urticaria pigmentosa (adult type).
And for tru-cut biopsy of liver we reported as bile duct proliferation and portal mast cell infiltration (Figure 7) (>40/40 High Power Field) staining with Giemsa and Toluidine blue (Figures 8 and 9 ).
On the basis of clinical presentation and laboratory investigations, we proposed our patient to be examined clinically for systemic mastocytosis. The diagnosis of systemic mastocytosis then confirmed by raised tryptase level.
Mastocytosis comprises a spectrum of related diseases in which there is an increase in mast cells in one or more organs. It usually occurs as a sporadic disease that is often transient and limited in children and progressive in adults. There may be symptoms, such as pruritus, related to the release of various products from these cells. The case we present here had also pruritus. Two KIT mutations at the 560 and 816 loci have been demonstrated to result in KIT auto activation, and they are believed to be responsible for increased mast cells arising originally from the bone marrow. Our patient had also kit positive young mast cell infiltration in bone marrow.
Figure 1: Young mast cell infiltration in bone marrow (H & E X100)
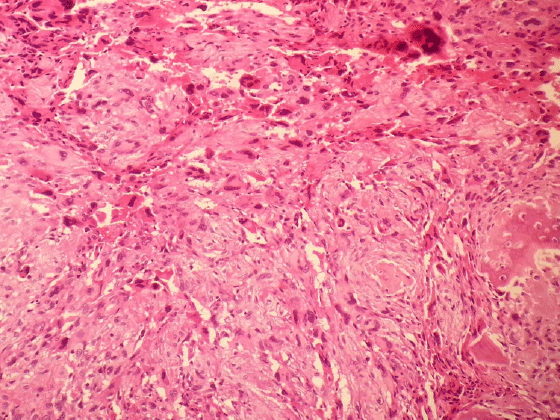
Figure 2: Young mast cell infiltration in bone marrow (H & E X200)
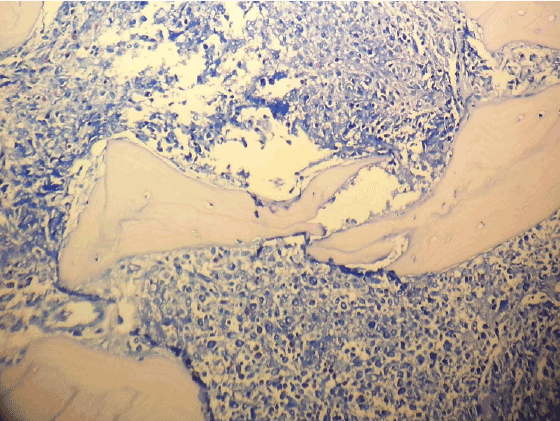
Figure 3: Mast cells in bone marrow (Giemsa X200)
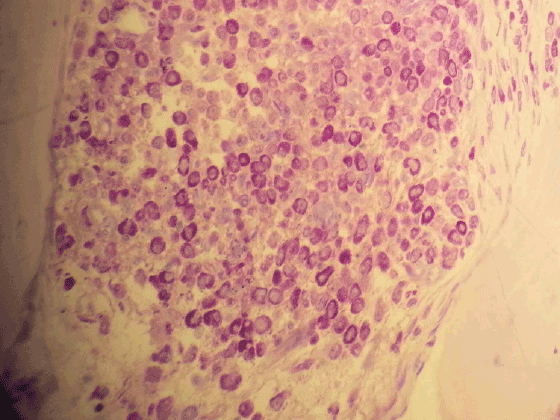
Figure 4: Mast cells with metachromatic granules (Toluidin Blue X400)
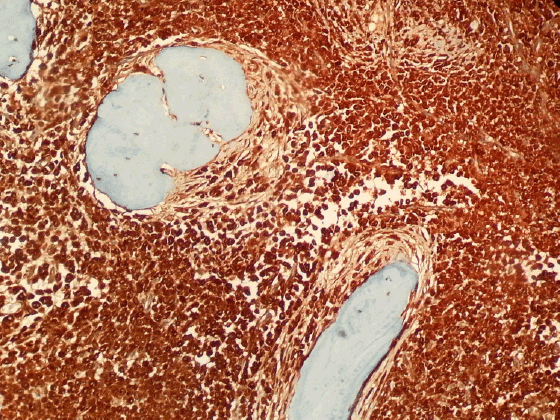
Figure 5: Mast cells in bone marrow (CD 117 X100)
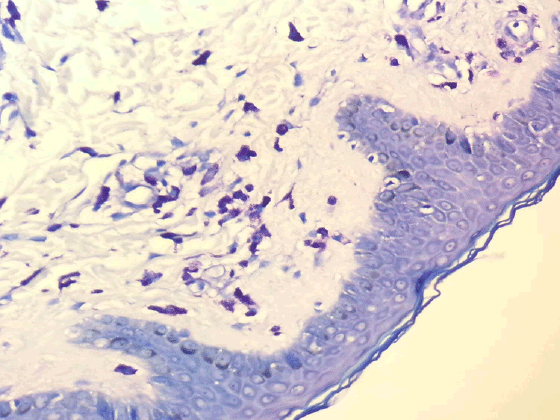
Figure 6: Mast cell infiltration in dermis (Toluidine Blue X200)
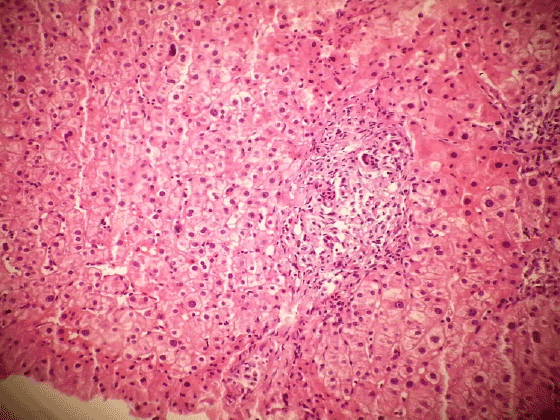
Figure 7: Portal mast cell infiltration (H & E X200)
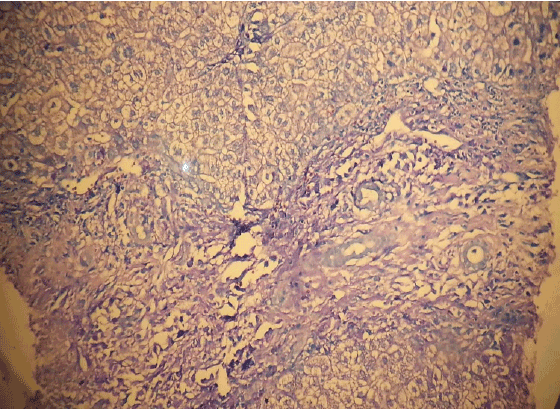
Figure 8: Portal mast cell infiltration in liver (Toluidine Blue X200)
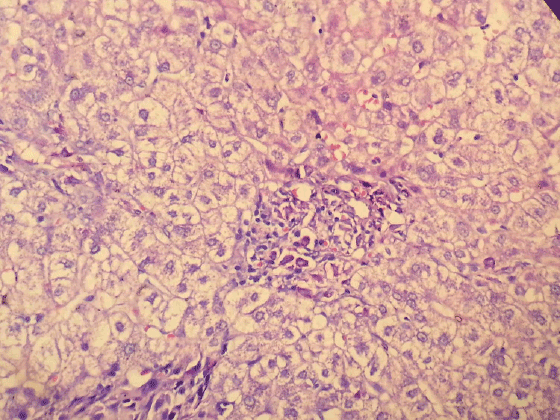
Figure 9: Hepatic mast cell infiltration with metachromatic granules (Toluidine Blue X400)
In systemic mastocytosis there is a proliferation of mast cells in various tissues apart from, or in addition to the skin. Systemic mastocytosis may develop in childhood cases of urticaria pigmentosa that persist beyond puberty, and in approximately 40% of adults with urticaria pigmentosa, usually of long standing.
Cutaneous lesions are most common on the trunk, although all skin areas, including mucous membranes, may be involved. Our patient had also cutaneous maculopapular lesions on the trunk.
The bone marrow is the tissue most frequently involved in systemic mastocytosis, followed by the liver, spleen, gastrointestinal tract, lymph nodes and rarely, other organs. Sometimes bone marrow is the only tissue involved besides the skin. The patient we present here had systemic disease; the skin, the liver, the spleen and the bone marrow were all involved.
Systemic mast cell disease without skin involvement is quite uncommon, and often difficult to diagnose. Flow cytometry on bone marrow samples, and the use of serum or marrow-blood tryptase levels may assist in making a diagnosis in indolent cases without cutaneous involvement. Fortunately our patient had skin involvement.
Systemic mastocytosis may progress to malignant mastocytosis and/ or mast cell leukemia. Various lymphoproliferative and myeloproliferative conditions may sometimes eventuate, particularly myelogenous leukemia. It has also been associated with a lymphocytic lymphoma. The hypereosinophilic syndrome is a rare association. Our patient is still alive .The disease didn’t progress. He used Ocladra (Kladribin) 2 mg per day.
Cutaneous lesions regress in approximately 10% of older patients who have systemic mastocytosis. In patients with an associated hematological condition, this regression may be accompanied by progression of the hematological disease. In patients with systemic mast cell disease without associated hematological disorders, the bone marrow mast cell burden, bone marrow eosinophilia, and serum alkaline phosphatase levels are of prognostic significance.
It has been claimed that up to one-third of individuals with systemic mastocytosis may progress to malignancy, but this seems unduly pessimistic in the light of other studies, one of which showed that the clinical course of systemic mastocytosis was stable over a period of 10 years in all those followed.
Download Provisional PDF Here
Aritcle Type: Case Report
Citation: Kimiloglu E, Ozcan Z, Erdogan N, Igdem AA, Mavis O, et al. (2015) Systemic Mastocytosis Presenting by Hepatosplenomegaly: Case Report. J Gastric Disord Ther 1(2): doi http://dx.doi. org/10.16966/2381-8689.108
Copyright:© 2015 Kimiloglu E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access