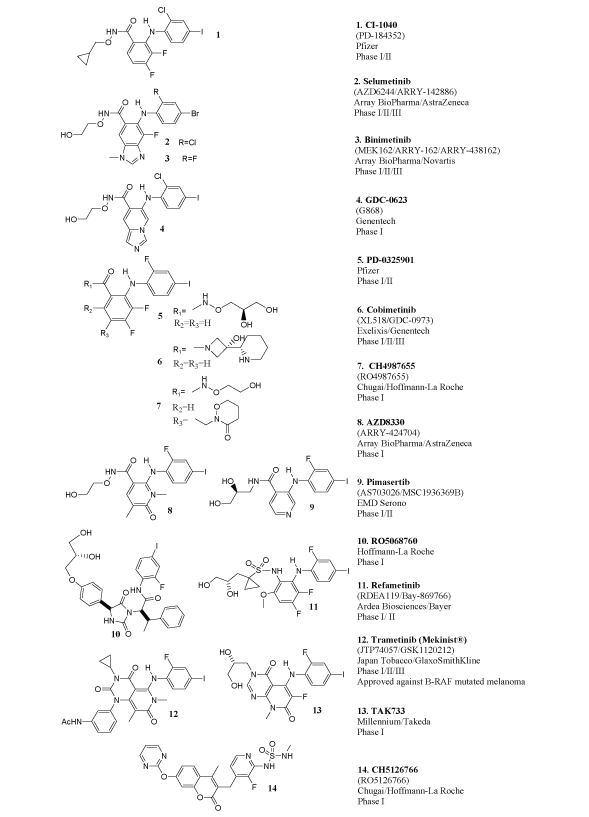
Figure 1: Allosteric MEK inhibitors that reached clinical trials

Allosteric MEK1/2 Inhibitors for the Treatment of Cancer: an Overview
Lee McDermott1* Chuchu Qin2
1Department of Pharmaceutical Sciences, University of Pittsburgh, 3501 Fifth Avenue, 10040 BST3, Pittsburgh PA 15216, USA*Corresponding author: Lee McDermott, Department of Pharmaceutical Sciences, University of Pittsburgh, 3501 Fifth Avenue, 10040 BST3, Pittsburgh PA 15216, USA, Tel: 412-648-9706; E-mail: lam179@pitt.edu
The RAS-RAF-MEK-ERK mitogen activated protein kinase (MAPK) pathway is a driver behind key cellular processes such as gene transcription, cellular proliferation, survival and motility and is aberrantly activated/dysregulated in many cancers. Mutations in RAS or RAF and/or increased signaling by receptor tyrosine kinases (RTK) associated with the cascade are frequently observed in many tumor settings and are linked to the dysregulation and aberrant activation of this pathway.
MEK is a central component of this highly important cascade and has a unique allosteric pocket adjacent to its ATP binding site. As such, there has been an intense pursuit of allosteric small molecule inhibitors of this kinase. This pursuit, to date, has resulted in the advancement of 16 compounds to clinical trials and the approval of trametinib (Mekinist®) for the treatment of melanoma. In this brief review we provide an overview of the RAF-RAS-MEK-ERK cascade, its cancer implications and brief account of the allosteric MEK inhibitors that reached the clinic.
Cancer, RAS/RAF/MEK/ERK, MEK, allosteric inhibitors, CI-1040/PD-184352, PD-0325901, Refametinib/RDEA119/BAY-869766, TAK733, Selumetinib/AZD6244/ARRY-142886, AZD8330/ARRY-424704, RO5068760, Cobimetinib/XL518/GDC-0973, CH4987655/RO4987655, CH5126766/RO5126766, GDC-0623/G868, ARRY-300, Trametinib/GSK112212/JTP-74057/Mekinist®, Pimasertib/AS703026/MSC1936369B, Binimetinib/MEK162/ARRY-438162, WX-554, crystal structures, clinical trials.
MEK is a dual specificity kinase and a central component in the RASRAF-MEK-ERK pathway, a well-characterized signaling cascade that is aberrantly activated in many tumors [1]. As MEK is a key component of this often dysregulated pathway, there have been many efforts to find inhibitors of this kinase as a potential treatment of cancer. The identification of small molecule inhibitors that bind MEK allosterically has been a major focus of these efforts. In this overview we provide a short discussion of the RAS-RAF-MEK-ERK signaling cascade and its involvement in cancer. We briefly discuss the current status and clinical findings of the allosteric MEK inhibitors that reached the clinic and the way these inhibitors bind to MEK.
The RAS-RAF-MEK-ERK cascade is a driver behind cellular processes such as gene transcription, cellular proliferation, survival and motility [1]. RAS, the first member of the RAS-RAF-MEK-ERK cascade, is a member of the super family of GTPases. RAS activation follows the activation of upstream receptor tyrosine kinases (RTKs) associated with the pathway and involves RAS complexation with cofactors Shc/Grb2/SOS. Inactive RAS then exchanges GDP with GTP and via a change in its conformation becomes activated. There are four RAS isoforms namely H-RAS, K-RAS (4A and 4B) and N-RAS [1,2]. RAF, the second link in the RAS-RAFMEK-ERK cascade, has three isoforms A-RAF, B-RAF and C-RAF (also known as RAF-1) [1,3,4]. RAF kinases are serine/threonine kinases and share similar structural characteristics. However, they differ in their ability to phosphorylate and activate MEK, the next kinase member of this signaling pathway, with B-RAF being a much more potent activator of MEK than C-RAF or A-RAF[1,3,4]. MEK has two isoforms, MEK1 and MEK2. Both MEK isoforms are highly homologous (80% overall similarity) and are activated by the RAF kinases through the phosphorylation of two serine residues in their activation loop (Ser218/Ser222 in MEK1 or the equivalent residues Ser222/Ser226 in MEK2) [1,5]. Upon activation, MEK1/2 phosphorylate their only known substrate, and the last member of the cascade, ERK [1,6]. There are two ERK isoforms, ERK1 and ERK2, and they are activated by MEK1/2 through phosphorylation on threonine and tyrosine residues (Thr202/Tyr204 in ERK1 or the equivalent Thr185/ Tyr187 in ERK2). The phosphorylated/activated ERK1/2 subsequently catalyzes the phosphorylation of multiple substrates in the cytosol and the nucleus that ultimately drive cellular proliferation, differentiation, survival and motility [1,6].
Since the RAS-RAF-MEK-ERK pathway plays a key role in the regulation of a multitude of critical cellular processes, it is not surprising that dysregulation and/or hyper activation of this pathway has been linked to cancer. Gain-of-function mutations, particularly in RAS and RAF, are important drivers in that regard. RAS mutations are observed in over 30% of all human cancers and among the RAS isoforms the frequency of K-RAS mutations is the highest (22%) while the frequency of N-RAS and H-RAS mutations is lower (8% and 2% respectively) [2]. B-RAF activating mutations have been observed in multiple cancer types such as colorectal carcinomas, breast cancer, non-small cell lung carcinoma, melanoma and others. Mutation frequency appears to be highest in melanoma with about 50-60% of melanomas harboring B-RAF mutations. C-RAF mutations have also been identified and are estimated to occur in approximately 1% of cancers[3,4,7]. MEK1/2 mutations in human tumors appear to be rare and there is no report in the literature suggesting prevalence of ERK1/2 activating mutations in human tumors [8-12]. Increased signaling through the RAS-RAF-MEK-ERK cascade may also be effected by the aberrant activation of upstream RTKs associated with the pathway as a result of increased ligand binding, increased dimerization or structural changes caused by genetic mutations in these RTKs [1,13].
Taking all of the above into account, it is evident that the RAS-RAFMEK-ERK cascade is a key pathway to target for the treatment of cancer. With respect to targeting specific components of the pathway, MEK has been viewed as a high value target [14]. There are multiple arguments in support of this assessment. MEK modulates only the activity of ERK, a kinase that modulates the activity of multiple substrates that are involved in cellular proliferation, survival and motility[1,6]. In this regard, MEK is a central node for the pathway and, as such, a point of strategic importance for intervention. Attempts to inhibit the pathway by interfering with RAS either directly or by inhibiting its farnesylation/translocation to the cell surface have not been particularly successful and, so far, the experience with RAF inhibitors in clinical use suggests that resistance builds quickly upon treatment with these agents [15,16]. As such, targeting MEK offers an alternative and complementary avenue to inhibit the cascade. Finally, as it will be shown in further detail below, MEK has a unique allosteric pocket adjacent to its ATP-binding site. This allows for the pursuit of highly selective allosteric inhibitors that, in principle, should be devoid of non-mechanism-related side effects and toxicities when compared to conventional ATP-competitive kinase inhibitors.
To date 16 allosteric small molecule MEK inhibitors have reached clinical trials. CI-1040 (PD-184352) was the first MEK1/2 inhibitor to be tested in humans [17]. Following CI-1040, the MEK1/2 inhibitors PD- 0325901, Selumetinib (AZD6244/ARRY-142886), AZD8330 (ARRY- 424704), Refametinib (RDEA119/Bay-869766), Cobimetinib (XL518/ GDC-0973), CH4987655 (RO4987655), CH5126766 (RO5126766), RO5068760, TAK733, Trametinib (GSK112012/JTP-74057/Mekinist®), Pimasertib (AS703026/MSC1936369B), Binimetinib (MEK162/ARRY- 438162), WX-554, ARRY-300 and GDC-0623 (G868) also advanced to clinical trials.
The structures of WX-554 and ARRY-300 have not been disclosed. For the remaining inhibitors the structures are shown in Figure 1. As evident from this figure, there are common structural motifs and a good degree of similarity among the majority of these inhibitors. Enzyme kinetics and crystal structure data confirm that these compounds, and their structural analogs, bind to MEK1/2 allosterically [18-30]. The available crystal structure data also suggest that, for the most part, the majority of these inhibitors bind similarly. For example, the x-ray structure of MEK1 in complex with PD-0325901, a CI-1040 analog, is available and shows that this compound, and the CI-1040 type of analogs in general, bind at an allosteric pocket adjacent to the ATP binding site via a common set of key interactions that involve the Lys97, Ser212, Val127 triad of residues (PDB: 3EQG, Figure 2A)[18,19]. In the MEK1 PD-0325901 cocrystal additional H-bonding interactions between the alcohol side-chain of PD-0325901 and the phosphate groups of the ADP present in the crystal structure are also seen but omitted in Figure 2A for clarity [19]. Cobimetinib, another compound from the broad CI-1040 class, binds to MEK1 in a similar fashion and via the identical key interactions with Lys97, Ser212 and Val127 with respect to the compound core (PDB code: 4AN2)[20]. With respect to the piperidine containing side-chain of cobimetinib there are additional interactions, specific to this compound, between the nitrogen and hydroxyl groups with Asp190 and the γ–phosphate of ATP [20].
The core of CH4987655 binds in an identical fashion (PDB code: 3ORN, Figure 2B) as PD-0325901 [21]. This is not surprising as the compound is part of the broader CI-1040 class of inhibitors. However, a unique feature in CH4987655 is its [1, 2] oxazinan-3-one side-ring. This ring resides in a pocket surrounded by Gly210, His188, Arg189, Asn221 and Met219 [21]. There is no apparent hydrogen bond between this side-ring and residues from the protein. The oxazinone moiety simply partially fills this pocket and effects a lateral displacement of the activation segment of the kinase [21]. Complexes of MEK1 with TAK733 (PDB code: 3PP1), refametinib (PDB: 3E8N) and selumetinib (PDB: 4U7Z), a sister compound to binimetinib, also show that these inhibitors bind in the same MEK allosteric site. Despite their apparently different functionalities and cores these compounds bind in a fashion similar to the one already described through key interactions with the Lys97, Ser212, and Val127 residue triad (Figures 2C-E) [23-25].
RO5068760 and CH5126766 are the most dissimilar with respect to structure when compared to the rest of the compounds shown in Figure 1. There is no published crystal structure available that elucidates the exact allosteric binding mode of RO5068760 with MEK. With respect to CH5126766, a cocrystal with MEK1 was recently disclosed and shows that this compound binds at the same MEK allosteric site (PDB: 3WIG, Figure 2F) [30]. However, the CH5126766 binding is differentiated as it does not involve the common key interactions with Val127 and Lys97 seen with other inhibitors. Only the interaction with Ser212 is maintained (Figure 2F). A key feature in the MEK1-CH5126766 cocrystal is that that the fluoropyridine sulfonamide moiety of CH5126766 projects into the cavity where the CH4987655 [1, 2] oxazinan-3-one side-ring resides, and similarly, effects a lateral displacement of the kinase activation segment. Unlike the oxazin-3-one ring of CH4987655, however, the fluoropyridine sulfonamide moiety of CH5126766 makes direct and/or water mediated hydrogen bonding interactions with Arg189, Asn221, and Ser222 (Figure 2F) [30]. CH5126766 has greater potency toward K-RAS mutant cells when compared to PD-0325901 and similar inhibitors. It has been proposed that the CH5126766 hydrogen bonding interaction with Ser222, which is a RAF phosphorylation site, in conjunction with the lateral displacement of the MEK activation segment caused by its binding, prevent MEK from being activated/phosphorylated by C-RAF in K-RAS mutant cells and this ultimately translates to the greater potency observed against this type of cells [30]. The conformational changes that result from the CH5126766 binding to MEK also result in the formation of tightly bound unphosphorylated-MEK/RAF unproductive complexes [30]. It is of interest to note that for compounds that bind through the Lys97, Val127, Ser212 triad, the interaction strength with Ser212 appears to be a determinant in their ability to effectively inhibit RAS mutated tumor cells. It has been proposed that a strong inhibitor Ser212 interaction confers a better stabilization and reduced flexibility of the MEK activation segment and that such rigidification prevents RAF from accessing its phosphorylation sites on MEK, stabilizes RAF unphosphorylated-MEK unproductive complexes and leads to greater potency in K-RAS mutant cells [31].
CI-1040 was the first MEK inhibitor that advanced to clinical trials [14,17]. This compound is potent (IC50 of 17 nM in the MEK enzyme assay) and showed activity in preclinical cancer models [32]. Its clinical development was halted in phase II, however, due to lack of efficacy [33]. High clearance that necessitated the use of very high doses (800 mg BID) and poor solubility that resulted in highly variable exposures have been cited as main reasons for the failure of CI-1040 in the clinic [14,33]. PD- 0325901 entered the clinic in 2004 as a replacement to CI-1040. It has improved pharmaceutical properties (solubility/stability) and greater in vitro (IC50 of 1 nM) and in vivo potency than CI-1040 [34]. Despite the improved properties and potency, an initial phase II study of this compound with non-small cell lung carcinoma (NSCLC) patients was prematurely terminated due to lack of efficacy and toxicity concerns[14,35]. Currently, PD-0325901 is being evaluated as a single agent in a phase II trial aimed to assess its efficacy against neurofibromatosis type-1 (NF1) and plexiform neurofibromas (NCT02096471). PD-0325901 is also under evaluation in combination studies with the PI3K/mTOR inhibitor PF-05212384 (NCT01347866), the EGFR inhibitor dacomitinib (NCT02039336) and the CDK4/6 inhibitor palbociclib (NCT02022982).
Figure 1: Allosteric MEK inhibitors that reached clinical trials
Selumetinib is also a potent allosteric MEK inhibitor (IC50 of 14 nM) with demonstrated activity in tumor cell lines with B-RAF and RAS mutations and several mouse xenograft models [36,37]. Selumetinib reached the clinic in 2004 where it was first studied in a phase I trial that involved 57 patients with advanced cancers. The results from this trial were published in 2008 and suggested that the compound was well tolerated at up to 100 mg BID dosing [38]. This initial trial was followed by a number of studies that aimed to assess selumetinib, as a single agent or in combination with other chemotherapeutics, in a number of cancer settings. In some of those trials end points were not met and/or the study report concluded that selumetinib offered no benefit [39-48]. Clinical activity that warrants further investigation was the conclusion of a recent phase II study report for the combination of selumetinib with paclitaxel in patients with K-RAS mutated NSCLC [49]. A similar conclusion was suggested in two reports from two phase II trials of selumetinib, as a single agent, in patients with low grade serous ovarian carcinoma and biliary cancer [50-52]. Another report from a phase II study of selumetinib in patients with metastatic thyroid cancer refractory to radioactive iodine treatment suggested that selumetinib, as a single agent, offers benefit and increases sensitivity to radioactive iodine treatment, especially in patients with N-RAS mutant tumors [53]. Interim results from an ongoing phase II trial of selumetinib versus temozolomide in patients with uveal melanoma also suggest that, as a single agent, selumetinib has greater activity than temozolomide in this chemotherapy resistant melanoma type [54]. Reports from phase II trials that studied the selumetinib/irinotecan combination as second line treatment in the K-RAS mutant colorectal cancer (CRC) setting and the selumetinib/erlotinib combination, in the chemotherapy refractory pancreatic cancer setting, concluded that these combinations warrant further exploration [55,56]. Preliminary activity was reported from two phase I trials of selumetinib, as a single agent, in children and young adults with neurofibromatosis or plexiform neurofibromas and in children with low grade gliomas [57,58]. Preliminary activity was also reported in phase I trials for combinations of selumetinib with the Akt inhibitor MK2206 and the EGFR inhibitor cetuximab [59,60]. At the time of this writing there were 40 recruiting and/or ongoing active clinical trials in the ClinicalTrials.gov registry in which selumetinib was listed as therapeutic intervention [61].
Cobimetinib has an in vitro IC50 in the MEK enzyme assay of less than 1 nM and has shown high potency and sustained growth inhibition of tumor xenografts in preclinical in vivo studies [20,62]. Data from phase I studies aimed to evaluate this compound either as a single agent or as a combination with the pan PI3K inhibitor GDC-0941 have been reported and suggested signs of efficacy and a manageable toxicity profile [63-65]. Data from a phase Ib study of cobimetinib in combination with the B-RAF inhibitor vemurafenib in patients with metastatic B-RAFV600 mutated melanoma also showed that this combination has activity, particularly in patients that are B-RAF-inhibitor-treatment naive [66]. Recently disclosed data from a phase III trial of the cobimetinib/vemurafenib combination, in the same cancer setting, showed an overall response rate of 68% and significant improvement in progression free survival [67]. On the basis of this data the FDA granted priority review to a new drug application (NDA) for the cobimetinib/vemurafenib combination for the treatment of patients with advanced B-RAFV600 mutated melanoma [68]. According to the ClinicalTrials.gov registry, at the time of this writing, there were 9 recruiting and active ongoing clinical trials with cobimetinib [69].
Refametinib exhibits MEK1/2 inhibition in the enzyme assay that is similar to that of CI-1040 (IC50 of 19-47 nM) and has demonstrated activity in multiple xenograft tumor models [23]. Refametinib has been involved in 6 completed phase I and/or II trials that studied this compound either as a monotherapy or in combination with gemcitabine, sorafenib and the PI3K inhibitor copanlisib [70]. A report from a phase I trial that evaluated refametinib, as monotherapy, in patients with advanced cancer concluded that this compound is well tolerated up to 100 mg QD and showed early signs of clinical benefit [71]. A similar conclusion with respect to tolerability and early signs of benefit was also reached in a recent report from a phase Ib trial of refametinib in combination with copanlisib [72]. Data from a combination study of refametinib with sorafenib in Asian patients with advanced hepatocellular carcinoma (HCC) showed that this combination has antitumor activity but high toxicity. Dose adjustments were needed for almost all patients that participated in that study [73]. A phase II study report of the refametinib/gemcitabine combination in advanced pancreatic cancer patients suggested that this combination has activity and there was a trend for better response rates in patients with K-RAS wildtype tumors [74]. At this time there are trials recruiting with the objective to study the combination of refametinib with sorafenib in RAS-mutated HCC (NCT01915602) and with the multikinase inhibitor regorafenib in patients with advanced or metastatic cancers (NCT02168777). There is also one ongoing refametinib/gemcitabine phase I combination trial focused solely on Asian patients (NCT01764828).

Figure 2: Binding mode and key interactions between MEK1 and PD-0325901 (A), CH4987655 (B), Refametinib (RDEA119) (C), Selumetinib (AZD6244) (D), TAK733 (E) and CH5126766 (F)
CH4987655, RO5068760, CH5126766, AZD8330 and TAK733 all have nanomolar range IC50 potency in MEK enzyme assays (5 nM, 25 nM, 160 nM, 7 nM and 3 nM respectively) and showed excellent activity in preclinical in vivo models [21,25,29,75,76]. These compounds have been profiled, as single agents, in phase I studies only. Data from the phase I trial of CH4987655 in patients with advanced metastatic tumors suggested that this compound has manageable toxicity and showed signs of benefit in 21% of the participants [77]. A report of the cohort expansion arm of this trial in patients with melanoma (B-RAF mutant and B-RAF wild-type), NSCLC (K-RAS mutant), and CRC (K-RAS mutant) was also recently published and concluded that CH4987655 showed early signs of activity in the melanoma and NSCLC arm of the study but not the CRC arm [78]. There is no phase I study of RO5068760 with cancer patients. The only available study with this compound is a phase I single ascending-dose study in healthy volunteers that suggested no clinically significant safety findings, moderate to high inter-subject variability with respect to pharmacokinetics and a rather moderate effect on the reduction of phosphorylated ERK levels in peripheral blood mononuclear cells of the study participants [79]. CH5126766 has been evaluated in two phase I trials one of which had only Japanese patients enrolled. Both studies reported a similar and manageable toxicity profile and preliminary antitumor activity [80,81]. The report from the only phase I trial of AZD8330 in patients with advanced cancers was published recently and suggested that the compound has manageable toxicity profile that is similar to other inhibitors of this class. The report also suggested that AZD8330 had clinical activity, defined as patients achieving partial response and stable disease for more than 3 months, in 26% of patients treated [82]. Results from the phase I trial of TAK733 suggested that there are hints of anti-tumor activity in patients with non-hematologic malignancies but high pharmacokinetic variability [83,84]. A planned phase I trial that aimed to study the combination of TAK733 with the aurora-A kinase inhibitor alisertib (NCT01613261) was withdrawn prior to patient recruitment [85].
Pimasertib has shown strong activity against tumor cell lines with oncogenic mutations or amplifications along the RAS-RAF-MEK-ERK pathway and tumor growth inhibition in mouse xenograft models [26,86]. According to the ClinicalTrials.gov registry, at the time of this writing, this agent was listed as therapeutic intervention in 7 active and recruiting studies [87]. Reports from a phase I study that evaluated pimasertib, as monotherapy, in patients with advanced tumors suggested a manageable toxicity profile and activity mostly in patients with B-RAF and N-RAS mutated melanomas [88,89]. Reports from a phase I trial of pimasertib in combination with PI3K/mTOR inhibitor SAR245409, in patients with advanced solid tumors, concluded that this combination is tolerated and shows signs of efficacy [90]. However, results from trials that aimed to evaluate pimasertib, as monotherapy, in patients with acute myelogenous leukemia (AML), the pimasertib/FOLFIRI combination, as second-line treatment for patients with K-RAS mutated metastatic CRC, and the pimasertib/gemcitabine combination, as a first line treatment, in patients with metastatic pancreatic (mPaCa), showed lack of efficacy and/or excessive toxicity [91-93].
Binimetinib has an IC50 of 12 nM against MEK and is efficacious in several xenograft tumor models [94,95]. According to the ClinicalTrials. gov registry, binimetinib is listed as a drug intervention in 23 recruiting and ongoing active trials [96]. A report from a phase I cohort expansion study of binimetinib, as monotherapy, in patients with biliary tract cancers suggested an acceptable safety profile and signs of clinical activity [97]. Reports from a phase II study with patients with N-RAS and B-RAF mutated melanoma suggested that binimetinib is active in both settings [98,99]. Preliminary reports of ongoing phase I/II trials of binimetinib in combination with the B-RAF inhibitor LGX818, the PI3K inhibitor BYL719 and the CDK4/6 inhibitor LEE011 suggest that these combinations are tolerated and showed preliminary activity [100-102].
There is not much known about the clinical activity of GDC-0623 (G868), Arry-300 and WX-554. According to the ClinicalTrials.gov registry, GDC-0623 (G868) has completed a phase I study in patients with locally advanced or metastatic tumors (NCT01106599) and Arry-300 has been profiled in a phase I study with healthy volunteers (NCT00828165). For either study, however, no data has been publicly disclosed to date. Up until recently, WX-544 was being investigated, as a single agent and in combination with the PI3K inhibitor WX-037, in two clinical trials (NCT01581060 and NCT01859351 respectively). However, these studies were terminated prematurely for business reasons and no data has been disclosed about them so far [103,104].
Trametinib is highly potent against MEK (IC50 of 0.92-3.4nM) [105]. In phase I trials in patients with solid tumors or lymphoma, trametinib, as a single agent, showed stable pathway inhibition, long half-life and clinical activity, particularly in patients with melanoma (confirmed response rate: 33% in B-RAF mutant; 10% in B-RAF wild-type)[106,107]. This activity was further confirmed in a phase II cohort study in previously treated metastatic, B-RAF mutated, melanoma patients. The study showed a 25% objective response rate with B-RAF-inhibitor-treatment naive patients but limited clinical activity in patients who had been treated with a B-RAF inhibitor and had developed resistant disease [108]. Following these results, a phase III trial in patients with B-RAF mutant melanoma that were B-RAF-treatment naive ensued. This study involved 322 patients and showed an objective response rate of 22% and progression free survival of 4.8 months for the trametinib arm of the study vs 8% and 1.5 months, respectively, for the control arm of the study. Survival rate at 6 months was also better in the trametinib arm of the study (81%) vs the standard therapy arm (67%) [109]. Based on the results of this study the US FDA approved trametinib (May 2013) for the treatment of patients with unresectable or metastatic melanoma with B-RAFV600E/K mutations [110]. A recent report from a phase III trial that assessed the efficacy of trametinib in combination with the approved B-RAF inhibitor dabrafenib (Tanfilar®) as a first line treatment for patients with B-RAF mutant melanoma concluded that this combination significantly improved overall survival in patients with metastatic melanoma with B-RAFV600E/K mutations and without increased overall toxicity [111]. This combination however does not confer significant benefit to patients that progressed while on B-RAFinhibitor monotherapy [112]. The US FDA granted accelerated approval to dabrafenib/trametinib combination in patients with unresectable or metastatic melanoma with B-RAFV600E/K mutations in January of 2014 [113]. Preliminary reports of phase I/II trials of trametinib in combination with dabrafenib and the EGFR inhibitor panitumomab in patients with B-RAF mutant CRC suggested early signs of benefit [114]. Also reports from recently concluded phase I combination studies of trametinib with other chemotherapeutic agents suggested that the trametinib/docetaxel or trametinib/pemetrexed combinations in patients with advanced NSCLC have clinical activity [115,116]. Recent reports pertaining to use of trametinib as monotherapy vs docetaxel in patients with advanced K-RAS mutant NSCLC or its use in combination with gemcitabine vs gemcitabine alone in patients with metastatic pancreatic cancer suggested no benefit in these settings [117,118]. No benefit, due to high toxicity, was concluded in a recently published phase I trial report with respect to the combination of trametinib with the mTOR inhibitor everolimus [119]. Studies with trametinib continue. At the time of this writing there were 77 recruiting and/or ongoing active clinical trials with trametinib listed in the ClinicalTrials.gov registry [120].
To date 16 allosteric MEK1/2 inhibitors have advanced into clinical trials and one of them, trametinib, has been approved by the US FDA for the treatment of B-RAFV600E/K mutated melanoma either as monotherapy or in combination with the B-RAF inhibitor dabrafenib [110,113].
The majority of allosteric MEK inhibitors that made it to the clinic have a good degree of similarity and common structural elements. Crystal structures for a number of these agents, or their analogs, with MEK1 are available and suggest that the majority of them, despite differences in cores and functionalities, follow a general binding motif that involves a set of common interactions with the Lys97, Ser212 and Val127 residues at an allosteric pocket adjacent to the ATP binding site.
Some MEK inhibitors are able to inhibit RAS mutant tumor cells better than others. The ability of MEK inhibitors to interact with the RAF phosphorylation sites of MEK and/or their ability to rigidify or laterally displace the kinase activation segment appear to be key in that regard. For instance, the ability of GDC-0623 to better inhibit K-RAS mutant cells than selumetinib, PD-0325901 and related agents is attributed to its strong H-bonding interaction with Ser212 that leads to less flexibility of the MEK activation segment and ultimately the inability of wild-type RAF to reach its phosphorylation sites, Ser218 and Ser222, on MEK [31]. Reports allude to the interaction of trametinib with Ser218, most likely via its acetyl group, as a possible reason for its ability to better inhibit the inactive un-phosphorylated form of MEK and to render it resistant to phosphorylation and activation by RAF in RAS mutant cells [28,30]. The greater effectiveness of CH5126766, versus that of PD-0325901 and similar compounds, in inhibiting the proliferation of K-RAS mutant cells, is attributed to its ability to prevent the C-RAF phosphorylation/ activation of MEK by interacting with Ser222 and by laterally displacing the MEK activation segment [30].
With respect to side effects, the commonly observed adverse events with MEK inhibitors include rash, diarrhea, fatigue, blurred vision and loss of visual acuity. CNS-related adverse events, elevation of creatine phosphokinase (CPK), retinal vein occlusion have also been observed [14]. Vision related adverse events appear to be unique to MEK inhibitors as opposed to adverse events seen with other kinase inhibitors.
Despite the importance of the RAS-RAF-MEK-ERK pathway in multiple cancer settings and the development of exquisitely potent MEK inhibitors, the clinical success with these agents so far has been narrow with respect to therapeutic window and scope. Primary resistance, due to feedback loops and cross talk with other pathways, or acquired resistance mechanisms offer an explanation for this assessment [14]. Combination regimens and the use of biomarkers that can identify patients that can benefit the most may be the answer to overcoming resistance and broadening the scope and success of treatment with MEK inhibitors. Toward that end, a number of studies aiming to identify biomarkers and/ or gene-expression signatures that may help predict response to MEK inhibitors were recently reported and clinical trials with MEK inhibitors in combination with other agents are in progress [121-124].
Download Provisional PDF Here
Article Type: Mini Review
Citation: McDermott L, Qin C (2015) Allosteric MEK1/2 Inhibitors for the Treatment of Cancer: an Overview. J Drug Res Dev 1 (1): http://dx.doi.org/10.16966/2470-1009.101
Copyright: © 2015 McDermott L et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access