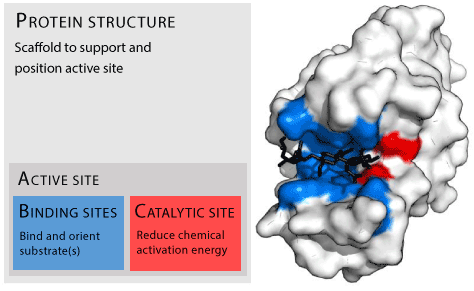
Figure 1: Protein structure “Enzyme structure” by Thomas Shafee - Own work. Licensed under CC BY-SA 4.0 via Wikimedia Commons.

GR Sridhar*
Endocrine and Diabetes Centre, Visakhapatnam, India*Corresponding author: GR Sridhar, Endocrine and Diabetes Centre, 15-12-15 Krishnanagar, Visakhapatnam 530 02, India, Tel: -891-2566301/2706385; E-mail: sridharvizag@gmail.com
Computational methods are being employed to predict active binding sites of enzymes. The principal reasons are that the pace of discovery of new proteins is increasing, outpacing the ability to characterize them in conventional biochemical and structural techniques; in addition advances in computational, structural and force data are used in an iterative manner to improve accuracy of active site prediction. From methods using amino acid and nucleotide sequences evidence is available that residues in the enzyme core are selected for stability while those at the surface, which are sites of protein interaction, trade off stability for ligand interactivity. In THEMATICS method the underlying concept is that active site residues involved in specific biochemical actions have predictable chemical properties which are identified using pH titrations of the enzyme activity. A more recent method uses a more comprehensive physico-chemical and electrical interaction map of amino acids at the surface of the protein which are likely to be sites of interactions with other proteins and chemicals. In summary, computational tools offer simulation and analytical opportunities to characterize, identify and modify proteins with appropriate characteristics to catalyze the specific biochemical reaction that is required. In this review the enzyme butyrylcholinesterase, involved in the expression of metabolic syndrome is presented, and recently available methods to predict protein structure, function and active sites are summarized.
Insulin resistance and type 2 diabetes are traditionally understood to result from a mismatch between insulin requirement and insulin supply [1]. Although essentially the concept still holds true, recent advances in biochemistry including bioinformatics are bringing to the fore newer players in the expression of these conditions [2]. While newer generation genomic methods have uncovered unsuspected proteins in the etiology, a long established protein, namely, butyrylcholinesterase is being studied in relation to the expression of insulin resistance and related conditions [3].
Butyrylcholinesterase (BChE) belongs to the esterase family of proteins, of which acetylcholinesterase is the most well known. The most well known role of BChE is the hydrolysis of succinylcholine, a muscle relaxant employed in general anesthesia. Individuals who had variant forms of the enzyme developed prolonged apnea with succinylcholine, and the enzyme was studied principally in terms of pharmacogenetics [3].
However, other roles for the enzyme were also studied. Evidence were presented accounting for its relation to dietary fat intake, obesity, insulin resistance, type 2 diabetes mellitus and Alzheimer’s disease [4]. Its role as a treatment for cocaine toxicity was also evaluated. Recent studies are bringing up the intriguing possibility of studying individuals harboring variant forms of the enzyme for their health outcomes [5,6].
Thus BChE is a protein in search of functions and may be considered as a prototype for the rapid identification of proteins by newer technologies. Therefore newer methods of assessing their structure and function are being developed. Bioinformatics tools are increasingly being used in this endeavour. This review provides an overview of bioinformatics and computational biological tools used in the characterization of such proteins.
It is well known that enzymes are protein molecules which perform a variety of cellular and regulatory functions by interacting with ligands at specific locations called ‘active sites.’ (Figure 1). The active site depends on a variety of factors, ranging from the sequence of amino acids (primary structure) to arrangement of protein chain in space and time (secondary, tertiary and quaternary structures). Interest in the structure and evolution of proteins in a broader perspective or ‘protein space from a global perspective’ [7] is gaining prominence. A network analysis of evolutionary path provides information about protein evolution and thereby development of proteins with unique properties.
The genome sequencing methods resulted in identification of large number of linear amino acid sequences without knowing the biological role of these proteins [8]. The active site is dynamic with interactions occurring with a variety of physical and chemical factors in the surroundings. With more data flooding than can be annotated, computational methods are employed in this area [9]. Predicting the active site leads to better understanding of a protein’s function and can be used for designing rationally that can modulate interaction of proteins by modifying their structure as well as to establish interactions among networks of proteins [10,11].
Figure 1: Protein structure “Enzyme structure” by Thomas Shafee - Own work. Licensed under CC BY-SA 4.0 via Wikimedia Commons.
The motivation for ‘reverse engineering’ of protein structure and function is the assumption that sequences of naturally occurring proteins are defined by selective pressure evolutionarily; the pressure is exerted by a balance of function and stability [12]. Studies comparing stability and solubility of proteins from their sequences have shown that core residues in proteins appear to follow the need for stability [13]. Efforts were made to use structure of known proteins and predict the amino acid sequences so that the information can be utilized to predict structure of new proteins whose amino acid sequences alone are known [14].
Many protein sequence design methods have been employed such as homology modeling in which attempts are made to match unknown to known, and then thread the sequence to a known backbone based on energy expression. Energy expression is in turn determined by stochastic and deterministic search algorithms.
Emil Fischer summarized the action of enzymes as follows: ‘In order to be able to act chemically on one another, an enzyme and its substrate must fit together like a lock and key.’ The original ‘lock and key’ concept has been revised and refined to describe the site where substrate and enzyme interact. The region of the enzyme containing the binding and catalytic sites is called the active site [15] (Figure 2). It usually occurs near or on the surface of the protein as a cleft for the substrate to fit. The active site is located in three dimensions, with amino acids or cofactors comprising binding and active sites. These arrangements are further affected by amino acids in the microenvironment of the active site.
Identification of binding and catalytic sites is a complex process, involving the use of substrate analogues, modification of amino acid side chains of enzymes, generation of fusion protein and by site-directed mutagenesis [16,17]. In essence, understanding enzyme action at the active site requires not only the identification of amino acids, but also the microenvironment in which they exist, and their spatial arrangement. Information about enzyme structure has been traditionally obtained by x-ray crystallography and NMR spectrometry.
Considering the complexity of the physical and chemical properties in which the interactions occur, computational prediction requires, besides the order of amino acids, the various physical and chemical interactions of atoms lining the active site gorge.
In this presentation, we review in silico methods that have been used to identify active sites of enzymes, with specific reference to butyrylcholinesterase which may be considered a protein with known structure but yet to be completely identified functions.
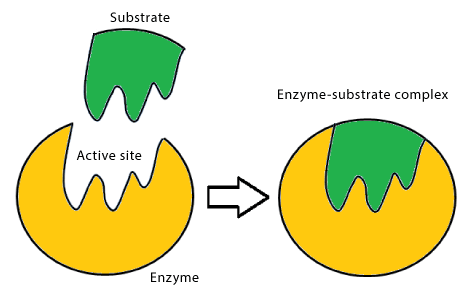
Figure 2: Lock and key model of enzyme action
Considering that proteins interact with other molecules to either enhance or inhibit biological functions, the key site is the ‘binding site’ through which ligands influence their action. Usually a limited number of key amino acids are located at the binding site. A number of computational methods may be employed to identify the active site. They are broadly classified into sequence based and structure based methods [18]. Other models involve statistical averaging of protein motion with conformations and calculation of free energy [19].
The basis of these methods is computational ability of functional inference from structural information that is available from omics technologies [20]. Structural methods can identify evolutionary relation among proteins. Since function is dependent on structure, structure can therefore give insight into a protein function. It must be recognized that these methods, sequence based as well as structure based do not directly give information about the biological role of proteins (Table 1).
One of the ways to predict active site is multiple alignments of related sequences that can build up consensus sequences of known families; domains, motifs or sites and Sequence alignments can give information on loops, families and function from conserved regions. The Cn3D software gives a consensus view of the related sequence, which is the best indicator of conservation. In addition, the amino acids also exhibit certain properties like conservation and hydrophobicity, which are indicative of active sites. In summary then, the active site can be predicted from the sequences.
The sequences of all organisms for a particular protein are gathered and a multiple sequence alignment is performed. Then the output of the multiple alignments is fed as the input to the prediction algorithm in chunks of 60 amino acids for each of the organism i.e an array of 60X number of organisms.
Prediction of active site can be achieved as follows: given the active site start position number and the sequences in 60X number of organisms the program first looks for hydrophilic amino acids and especially for histidine in the human sequence, which is rarely present in the protein except for the active site.
On finding the hydrophilic amino acid, the program checks the consensus sequence, which is obtained from the sequences of other organisms. If the same amino acid is not found then the consensus sequence is checked for a nearer hydrophilic amino acid based on the properties. If that fails then the program moves onto the next amino acid in the human sequence. If the same amino acid is found then conservation of the amino acid in the subsequent sequences are observed by searching for that amino acid or a nearer amino acid in the sequences below which would add to the accuracy with which one can predict an active site.
Two factors can be obtained in this program -- accuracy and the position of the active site. Accuracy is measured by taking into account factors such as conservation of protein in other organisms while giving priority to the amino acids (e.g. histidine), which have greater probability of being a part of active site. Accuracy increases if the amino acid is conserved in all sequences.

Table 1: Overview of available methods*
Secondly, the position of the active site can be predicted since the start number is given by the user. The position where there is possibility is checked and added to the start number of the sequence which predicts the position of the active site in the protein.
The assumption is that those amino acids with predictions of accuracy 90% and above is indicative of active sites.
THEMATICS is a computational procedure to identify active sites in proteins, in which the 3-dimensional structure of a protein alone is taken as input from the Protein Data Bank Website. This method identifies reactive sites, including residues involved in catalysis [21] (Table 3).
This technique utilizes Deep View-Swiss Pdb Viewer method to obtain the electrical potential function of the protein, followed by calculation of the predicted titration curves for all of the ionisable residues in the protein structure. The shapes of the predicted titration curves are analysed to identify those residues with elongated, non-sigmoidal titration behavior. The underlying basis for this method is that some amino acids have ionisable side chains, ie Lysine, Arginine, Histidine, Tyrosine, Asparate, and Glutamate, which frequently occur in active sites of enzymes.
In proteins, active sites usually comprise hydrophobic pockets that involve side-chain atoms. Methods which use interaction energies between the receptor and different probes to locate energetically favorable sites are complex. They require the assignment of proton locations and partial charges to the receptor atoms. While van der Waals energies can indicate sterically available regions, the long-range nature of electrostatic potentials makes the interpretation of energy levels difficult.
The purpose of the Site Finder application in MOE is to calculate possible active sites in a receptor from the 3D atomic coordinates of the receptor.
THEMATICS offers the possibility of identifying active sites of enzymes when its structure is known, even in the absence of biochemical data. The underlying concept is that active site residues involved in specific biochemical actions have predictable chemical properties which are identified by using pH titrations of the enzyme activity.
This computational method can identify active site of protein even without biochemical information, and depends only on a threedimensional structure. The underlying concept is that the ‘pH dependent electrical potential caused by ionizable residues … perturbs the substrate and the catalytically active residues, so that efficient … proton transfer is enabled between them over the desired pH range’ [21].
Computational methods, mostly sequence based, were developed to predict interface sites at differing resolution: at the entire domain, a sequence or at the level of each amino acid [11]. In general, interface residues are predicted by their known characteristics, evolutionary conservation of residues and by residue energy distribution on protein surfaces [10]. Known characteristics include the fact that protein interfaces occur at the planar and accessible paths.
|
Table 3: Methods to identify binding sites
Using correlated mutations, residues close to interaction site are expected to mutate simultaneously during evolution, in contrast to coevolution approach where simultaneously mutations are considered at two interacting partners rather than a single protein (Table 3) [11].
Sequence-based methods depend on the genomic context, i.e. the primary structure, on gene order conservation and on domain homologues [11].
It should be emphasized that such predictive methods need an iterative experimental characterization and production, even though it assures a ‘comprehensive measure of the compatibility of a sequence with a structure’ [14].
In addition, a variety of statistical predictive methods are employed using neural networks and machine learning techniques [14,22-24]. Often a combination of methods is used, involving neural networks, docking and superimposition [25]. Such predictive methods sometimes use well known phage display libraries (e.g. SiteLight method) [11].
Given the fact that most of the amino acid sequences in a protein are optimized for structural stability and are often buried within the structure, attempts were made to perform a more comprehensive physicochemical and electrical interaction map of amino acids at the surface of the protein which are likely to be sites of interactions with other proteins and chemicals [26].
In their original study, Chakrabarti et al. chose a set of 10 amino acid residues that form essential contacts to the ligand for sequence optimization. The essential contacts were identified by residues necessary for hydrogen-bonds, salt bridges, van der Waals forces or hydrophobic contacts. The latter was obtained using mutagenesis data and by multiple sequence alignments [26]. The three principal steps in the procedure included determining the lowest-energy protein structure for each residue, following sampling of side-chain conformation and calculation of ligand-binding affinity for each.
By this method, it was observed that most binding-site amino acids were ‘optimized … for simple scoring functions based on ligand-binding affinity, under the constraint that residues involved in catalysis are restricted to catalytically favourable conformations’ [26].
Machine learning algorithms have been employed for prediction of functional site where ligand-protein interaction occurs. In a comparative study, using WEKA software package, sequential minimal selection algorithm was found to correctly predict catalytic residues in the majority of enzymes utilizing seven attributes [27]. There is requirement for proper choice of machine learning algorithm along with optimal attribute sets for the algorithm.
Another hybrid method to predict binding sites uses computational methods that can identify surface pockets (binding sites) and the key amino acid residues in them. A prediction model employing Bayesian Monte Carlo method was found to reconstruct history of binding surfaces in evolutionary terms [28]. In view of the early sequence methods which required 60-70% identity between known and comparative protein, the hybrid methods make it possible to design of pharmacological agents which interact at these sites.
Spatial statistical methods can also be employed to identify conserved residues of homologous amino acids. An index has been devised to select those specific sequences that best help identify conserved residues of active sites [29]. The index, which shows the degree of conserved residue clustering on protein tertiary structure, was able to improve prediction of functional regions when both were integrated. This combined method of spatial statistics applied to protein analysis resulted in improved predictive ability (Table 2).
Methods are being devised to predict ligand binding sites for those proteins without experimentally solved structures. FINDSITE is used to predict binding site and functional annotation by looking for similar groups of weakly homologous templates which are identified by a threading procedure [30]. This method depends for its ligand-binding site identification by similarity of biding-sites among superimposed groups of templates; these are in turn identified by threading. Brylinski and Skolnick (ref just above) showed that FINDSITE is accurate in assigning molecular function.
SITEHOUND is another toolkit useful for the identification of protein ligand-binding sites. Using protein structure which is fed in PDB file, the Molecular Interaction Fields obtained are in turn input into SITEHOUND. The latter gives out a list of possible sites where binding of ligands can occur [31]. This freely available toolset which can be used on various computer platforms makes it possible to identify ligand binding through calculation of molecular interaction (Download of executables available from http://sitehound.sanchezlab.org/download.html)
Newer statistical methods are devised to predict catalytic residues at the ligand binding active site utilizing statistical prediction methods. While early predictive models used conservation of sequences in a family, others employed data from solved 3 dimensional structures with analysis of features such as geometry, electrostatistics, energetics and chemical properties. In order to improve prediction accuracy, even combining structure and sequence methods, the method described by Sankararaman et al. DISCERN improved the precision of ligand binding catalytic residues over previously existing methods [32]. DISCERN employed a confluence of conservation information, 3 dimensional structural data and a statistical regularization process.
Prediction of ligand-binding sites and catalytic residues within them provide functional information about proteins derived from sequence data, as well as pave way for chemical moieties which can interact at the site and have potential pharmacological roles.
| Energy based |
|
Table 2: Hybrid methods
In an exploration of sequence space that was associated with a given protein attribute, all-atom models were used along with a physical energy function [16,33]. It was found that the volume of sequence space was defined by protein length, protein topology and stability of protein fold. Folding free energy function selects naturally available protein sequences in the core but not on the surface [17,34]. This is utilized to search sequence space for sequences that satisfy stability constraints for known protein structures, which are then compared with naturally occurring counterparts. Information about native sequence appears to be ‘encoded’ in the backbone structure of protein [34].
Core amino acid sequences were on an average 50% identical to their native counterparts. Surface region identity scores were much lower than core amino acid sequences [34]. Newer methods such as Hidden Markov models are being explored for better identification of appropriate sequences.
It has been suggested that sequences on the surface of enzymes may have been selected at least in part, for mediating interactions at the expense of protein stability. Amino acids at the surface were therefore, optimized not for protein stability, but for functional reasons [34].
In summary, there has been synergistic use of data and methods obtained from different sources to give meaningful biological solutions to questions surrounding the structure and function of proteins; that are available in geometric progression, faster than can be characterized by hitherto traditional methods. Computational tools offer simulation and analytical opportunities to characterize, identify and modify proteins with appropriate characteristics to catalyze the specific biochemical reaction that is required.
The ultimate purpose of these computational tools is to design proteins with desirable properties that can interact with ligands of interest [35].
EvoDesign algorithm provides a method to identify new protein sequences containing the desirable sequence and structure for welldefined biological purposes. It is a web server where optimal protein sequences are designed with required scaffolds, sequence based methods that can analyze fold ability and thereby functional characters [36]. (Tables 4 and 5). It is available at the URL: http://zhanglab.ccmb.med.umich.edu/ EvoDesign.
Observing that optimization of scoring function can computationally predict residues in enzyme sites, an extension to other protein site predictions was carried out. A variety of steps were needed, including impact of hydrogen-bonding networks needed for catalysis, and sequence optimization accuracy with their incorporation into catalytic constraints. Simultaneous selection pressure was shown to determine the sequences found at active sites. Selected enzymes of the group kinases showed accuracy of sequence for topological similarity [37]. The authors conclude that the ‘designability of an enzyme active site’ could be employed as a metric in the search for proteins required for desired actions.
EvoDesign All atom force field |
Table 4: Methods to design new proteins
| Global root mean derivation CAMEO system EvoDesign |
Table 5: Quality assessment methods and web-based tools
While low-resolution prediction models for active sites are available, problems arise in achieving high-resolution prediction, on which further chemical derivations can be carried out. Procedures for bringing about structures closer to the native state were employed using energy function. It was shown that a ‘physics-based all-atom potential’ could have the ability to produce refined structural predictions [38]. Coarse-grained prediction is dependent on efficient search algorithms. But energy function should be overlaid for the native state of protein (Table 4).
With the availability of various computational protein prediction techniques, a comparison of quality among them is essential. Many variables exist, with the quality of template being the most important. In addition proteins that are related by evolution have different structures though the sequences are similar. The global root mean square derivation is combined with other parameters for predicting quality of protein tertiary structures. A step wise application improved the performance (Table 5) [39].
There has been a continual assessment of existing protein structure homology modeling which is now the standard technique whereby 3 dimensional models of proteins are designed without the availability of experimental structures. Expert system for SWISS-MODEL uses model quality estimation for selection of most suitable template. Its function is subject to evaluation by CAMEO system [40]. It is available at http:// swissmodel.expassy.org/
With the ability to provide in silico models of unique designer proteins, optimization methods are being described. Based on the transition state theory of catalysis and graph-theoretical model, an optimization model was developed for selecting binding sequence in enzyme design using computational methods. Identification of native binding sites was shown to be possible by taking into considering catalytic geometrical constrains and substrate structural motifs. [41]. Such information provides the ability to design novel enzymes with specified enzymatic properties.
With a large number of sequences being identified by omics technologies in diabetes, there is potential for widespread use of such in silico methods to annotate the putative structure and function of proteins: e.g in studying genes following an invoked inflammatory stimulus [42]. Besides known such as those involved in recruitment and activation of lymphocytes, adhesion molecules, antioxidants, several other genes with novel or unknown function were identified. Other genes such as JAZF1 (juxtaposed with another zinc finger gene 1), CDC123, CAMK1D, TSPAN8 are associated diabetes mellitus [43].
Similarly placental genes in women with gestational diabetes identified a large dataset of genes involved in stress-activated and inflammatory responses, with upregulation of interleukins, leptin, and tumor necrosis factor-α receptors and their downstream molecular adaptors. More than 100 genes were reported to be ‘unclassified’ and can be a potential source of structural and functional annotation [44].
I thank Dr G Lakshmi MD for her help in the preparation of the revised manuscript.
Download Provisional PDF Here
Article Type: Review Article
Citation: Sridhar GR (2015) Modulator of Diabetes and Metabolic Syndrome: Silent Proteins, newer Insights. J Diabetes Res Ther 1(2): doi http://dx.doi. org/10.16966/2380-5544.107
Copyright: © 2015 Sridhar GR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access