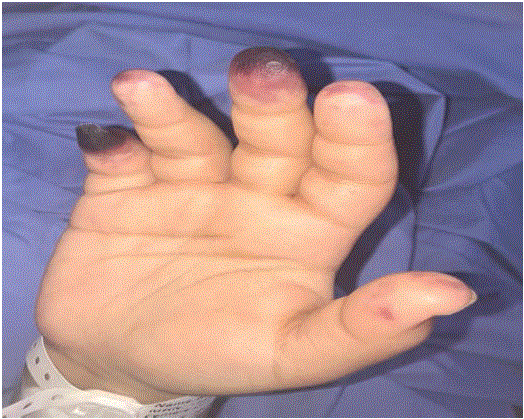
Figure 1: Shows discoloration indicates necrosis of the fingertips.
Abdulmohsen Alyousef1 Mohja Alshehri2*
1Department of Internal Medicine, King Abdulaziz national guard hospital, Al Ahsa, Kingdom of Saudi Arabia*Corresponding author: Mohja Alshehri, Department of Dermatology, King Fahd university hospital, khobar, Kingdom of Saudi Arabia, E-mail: mdshehri@outlook.sa
Introduction: Systemic Lupus Erythematous is an autoimmune connective disease that has vascular involvement and can manifest as acute/subacute inflammation as antiphospholipid syndrome. It can result in a predisposition for vascular thromboses and pregnancy complications.
Clinical manifestation: 20 years old female presented with fever for two weeks with bluish discoloration of the fingertips while her lower limbs swelled and gained a violaceous rash in a netlike pattern. On examination, she was vitally unstable. Bluish discoloration of the fingertips and lower limbs had bilateral retiform purport and lived racemose. The histopathological examination of the lesions showed both small and medium vessels vasculitis with perivascular inflammation and panniculitis. The patient was treated with analgesics, immunosuppressant (Rituximab), and steroid.
Conclusion: The histopathology examination is the key to make the diagnosis and the mainstay treatment is immunosuppressant.
Systemic Lupus Erythematous (SLE), is an autoimmune connective disease [1]. It has a heterogeneous manifestation of vascular involvement as the most common symptom. Vasculopathy can manifest as acute/subacute inflammation as antiphospholipid syndrome, lupus vasculitis [2]. Other related clinical diseases include thrombocytopenia with thrombotic purpura, venous thrombosis, antiphospholipid syndrome, and urticaria vasculitis. Such conditions are found to be between 11% to 37% of SLE patients [3].
The involvement of medium-sized vessels can cause obstruction. Livedoid vasculopathy, a certain form of systemic vasculitis, can cause an uneven perfusion of the lower limbs and manifest with chronic ulceration [4]. The presence of specific antibodies targeting phospholipid-binding plasma proteins is an autoimmune condition known as antiphospholipid syndrome [5]. It can result in a predisposition for vascular thromboses and pregnancy complications [6].
Although the true pathophysiology of the condition is not yet understood, it is believed to be a result of the natural course of the autoimmune disease and hypercoagulable state found [7]. The histological findings of the involved vessels show intravascular fibrin, segmental hyalinization, and endothelial proliferation which favor the thrombosis formation [8]. The presence of immunoreactions in the vessel wall and circulating immune complexes, such as a factor related to rheumatoid, are in favor of its immunological component [9].
It is important to differentiate lupus vasculitis from true vasculitis as each one involves a separate management plan. To do so, one should examine the patient’s vessel wall. If there is an inflammatory infiltration of the vessel wall, along with an absence of fibrinoid necrosis, the condition is livedoid vasculopathy. These key factors differentiate livedoid vasculopathy from true vasculitis [2] and by checking for these signs; the doctor can know which condition they must treat for the patient.
A clinical case will be examined in this paper, involving an unusual case of lupus vasculitis in a systemic lupus patient.
The case of a 20-year-old female (later referred to as patient T), with a continuous fever that began 2 weeks before the examination, is presented with fever and progressive development of painful swelling in the patient’s hands, with a bluish discoloration of the fingertips (Figure 1), along with a swelling of their lower limbs accompanied by a persistent violaceous rash in a netlike pattern.
Figure 1: Shows discoloration indicates necrosis of the fingertips.
The patient was presented to the emergency room. Her fever durinal variation wasn’t relieved by analgesics, the use of analgesics was neither measured nor documented, and she had sought medical help elsewhere around the time and was believed to have respiratory infection, because of her sore throat, and thus given antibiotics. The symptoms, however, did not improve.
The patient stated that, around the same time she got a sore throat, her upper and lower limbs started swelling. The swelling of the upper limbs was described as painful and to have increased in size over the two weeks before her examination. The patient’s fingertips had bluish discoloration while her lower limbs swelled and gained a violaceous rash in a netlike pattern (Figure 2).
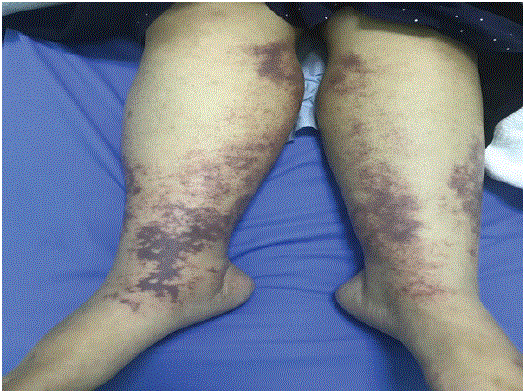
Figure 2: The lower limbs show violaceous rash in a netlike pattern.
After inspecting the patient’s past medical history, it was found that she got admitted into a hospital, nine months before the mentioned symptoms came to show, for an investigation of pyrexia of an unknown origin that did not conclude anything. A review of her system, however, came up positive with Raynaud phenomenon.
On physical examination: The patient was conscious, alert and oriented to time, place, and person. With this being said, however, she was quite ill-looking and pale. Regarding vital signs, the patient was tachycardia with a pulse of 125, fever of 38.1 orally, a respiratory rate of 20, blood pressure of 129/64, and 100% O2 saturation in room air. The fingertips of both hands had a blue discoloration while the lower limbs had bilateral retiform purpura and lived racemose involving the shaft extended to below the knees (Figure 3).
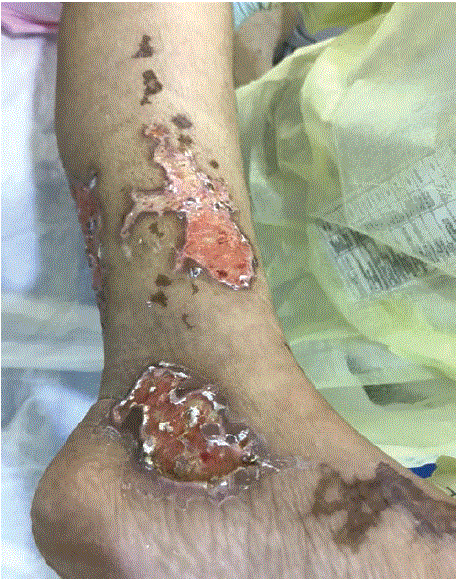
Figure 3: Follow up after 3 weeks from the start of treatment, vascular compromised results in tissue necrosis but also wound healing taking place.
The hospital course: The patient was then admitted into the medical ward by the internal medicine team. The rheumatology and dermatology teams were consulted. Basic laboratory investigations showed a white blood cell count of normal value with a differential of neutrophils at 2.5 k/UI, lymphocytes 17 k/IU, monophils 0.3 k/IU, eosinophils 0 k/IU and basophils of 0 k/IU. Low hemoglobulin was at 7.7 g/dl, Normal red blood cell count and normal platelets count. The estimated sedimentation rate was normal, C-reactive rate protein 2.8 mg/dl. The blood and urine culture came up as negative. PT, PTT, and INR of normal values. The liver function test and renal function test gave normal results and the patient’s Hepatitis profile came as negative. Serological laboratory results showed Complement C4: 2, Complement C3: 24, Ds DNA antibody 254, ANA value was high of >1280, Anti-smith antibody value was high of >693, β2 glycoprotein1 IgG 23.7, Anti-Sjogren antibody high of 834.3, anti-CCP antibody high 58.7, B2 glycoprotein1 IgM negative and an MPO <3.2.
A skin biopsy, with the sample taken from the involved lesions, showed both small and medium vessels vasculitis with perivascular inflammation and panniculitis (Figures 4 and 5).
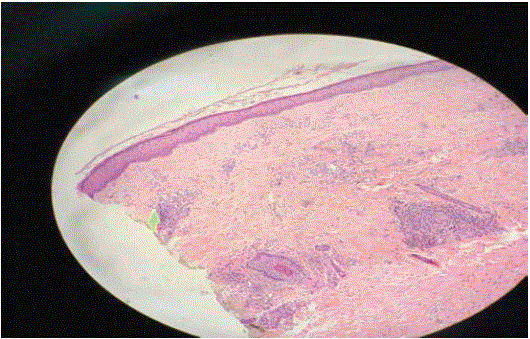
Figure 4: Histopathological picture of the lesion and shows both small and medium vessels vasculitis with perivascular inflammation and panniculitis.
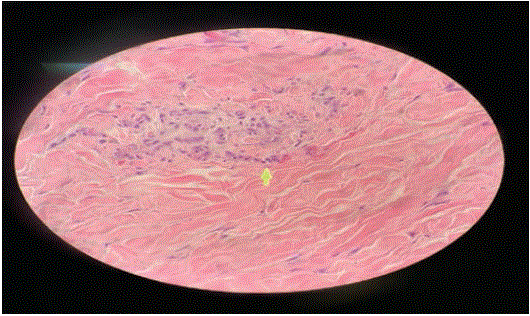
Figure 5: Histopathological picture of the lesion and shows both small and medium vessels vasculitis with perivascular inflammation and panniculitis.
The differential diagnoses: Anti-phospholipid syndrome associated with SLE versus Vasculitis secondary to autoimmune connective tissue disease. The treatment plan of this patient was to start on Rituximab 1000 mg single dose, Aspirin 100 mg po od, Paracetamol 1 g IV, Clexane 60 mg subcutaneous q12h, Tramadol 50 mg IV PRN q12h, Prednisolone 70 mg tab OP OD, Calcium Carbonate 600 mg OP OD, Cellecept 500 mg PO BID and betamethasone topical cream. Close observation was maintained to adjust the immunosuppressant.
Systemic Lupus Erythematosus (SLE) vasculitis prevalence for the patient was believed to be between 11% and 36%. However, its actual prevalence may have been underestimated because it is sometimes asymptomatic [10]. Vasculopathy with a measurement of 60% or higher involves leukocytoclastic inflammation, 30% involves cryoglobulinemia and 6% involves systemic vasculitis. Other vasculopathy issues, including antiphospholipid syndrome, report less than 5% [11-13]. Extracutaneous vasculitis is a rare manifestation of SLE [2].
There are several cases of SLE with antiphospholipid syndrome and small-vessel vasculitis. An example of one is found in a 60-yearold woman presented with edema of the left leg and positive antinuclear antibodies. Laboratory data revealed lymphocytopenia and hypocomplementemia while the anti-DNA and antiphospholipid antibodies came up as positive. Contrast-enhanced Computed Tomography (CT) showed deep vein thrombosis in her right leg and a diagnosis of SLE and antiphospholipid syndrome was then made based on these findings. Histological analysis of the muscle biopsy showed necrotizing vasculitis and thus confirmed a finding of lupus vasculitis. With the condition established, the patient was eventually prescribed prednisolone (0.6 mg/kg/day) and hydroxychloroquine. She improved clinically and the edema reduced.
Another case involved a 63-year-old woman diagnosed with atherosclerotic peripheral vascular disease and lupus anticoagulant. The patient developed ischemia of the right lower limb and thus required progressive amputations. Histopathological analysis showed inflammatory vasculitis in multiple arteries while her serum showed high titer anticardiolipin antibodies. Treatment with high-dose corticosteroids was then done which reversed the ischemic process.
Some patients test positive for antiphospholipid antibodies. They manifest with cutaneous lesions and/or gangrene as they are found to have underlying thrombosis and/or vasculitis [14]. Patient T was diagnosed with antiphospholipid syndrome, confirmed by clinical manifestations and laboratory results. Vasculitis was also confirmed by the results of an undertaken skin biopsy.
Abnormal vasospasm of the digital arteries, precapillary arterioles, and arteriovenous, in response to the cold and various other stimuli, is a rare complication of SLE and can cause a manifestation of digital discoloration known as Raynaud’s phenomenon. Digital ulceration is a rare complication [15] however not unheard of. A past case featuring a 45-year-old female, with a 10-year history of SLE accompanied by Raynaud’s phenomenon, can be found where the patient had severe pain, cyanosis and digital ulcers on her fingers [16].
The most common cause of digital gangrene, associated with SLE, is that of antiphospholipid antibody syndrome (AAS) while SLE is the leading cause of secondary AAS [15]. The treatment plan, for the 45-year old patient, involved iloprost infusion, bosentan, and immunosuppressive therapy (cyclophosphamide and pulse methylprednisolone). Six months of taking immunosuppressive oral cyclophosphamide and a combination of the calcium channel blocker nifedipine and low-dose ASA as an anti-platelet agent [15] was a key part of the process.
Patient T also had digital gangrene, and this was most probably caused by vasculopathy, related to Antiphospholipid syndrome. The treatment plan of a case report from Sudan published in 2019, which showed the effectiveness of rituximab in treating antiphospholipid syndrome was found. The reported patient had several miscarriages and one premature cesarean section. Three days after taking rituximab, the patient’s condition rapidly deteriorated and they developed anuria, high blood pressure (170/110), generalized body swelling, and signs of pulmonary edema. Lupus anticoagulant, anti-cardiolipin antibodies, and β2 -Glycoprotein were negative. The treatment consisted of prednisolone 40 mg, furosemide 4 mg, amlodipine 5 mg, calcium carbonate 500 mg. The patient also received two doses of immunoglobulin; however, this part of the treatment was stopped because she developed an allergic reaction to the substance. After such an incident, an infusion of Rituximab was then given to the patient, with 500 mg per one dose, to be taken for 14 days. Following this treatment, the patient’s general condition was improved [17].
Patient T was then also treated with Rituximab, of 1000 mg per single dose, along with Aspirin 100 mg po od, Paracetamol 1 g IV, Clexane 60 mg sub-cut q12h, Tramal 50 mg IV PRN q12h, Prednisolone 70 mg PO, Calcium Carbonate 600 mg OP OD, Cellecept 500 mg PO BID and betamethasone topical cream. After 3 weeks, her condition began to show improvement and the healing of a wound started to take place. However, it was found in the literature that the use of hematopoietic stem cell treatment was used in some cases for the treatment of vasculitis as it was able to demonstrate the presence of elevated numbers of circulating endothelial cells [18].
As found in this study, skin can show the first sign of the manifestation of Anti-phospholipid syndrome. This being said, the presence of any underlying rheumatological diseases should be ruled out in a patient, with retiform purpura, before the start of any immunosuppressive therapy. The spectrum of autoimmune disorders is wide-ranging from single organ involvement to systemic disease. The disease process is usually prolonged and is generally associated with significant mortality and morbidity however it is concluded that the mainstay of the treatment is to maintain immunosuppression.
Download Provisional PDF Here
Article Type: CASE REPORT
Citation: Alyousef A, Alshehri M (2020) Vasculitis and Antiphospholipid Syndrome in a Systemic Lupus Patient. Autoimmun Infec Dis 3(1): dx.doi.org/10.16966/2470-1025.126
Copyright: © 2020 Alyousef A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access