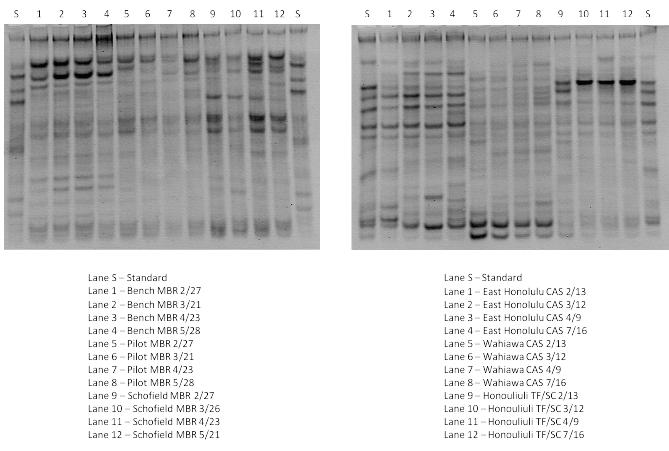
Figure 1: DGGE fingerprints of PCR amplified 16S rRNA fragments extracted from samples of mixed liquor from bench, pilot, and full-scale (Schofield) MBRs; and from East Honolulu CAS, Wahiawa CAS, and Honouliuli TF/SC.

He Xu-Sadri1 Krishna M Lamichhane2 Roger Babcock3*
1HDR Inc, 1132 Bishop Street, Suite 1200, Honolulu, HI 96813, USA*Corresponding author: Roger Babcock, Associate Professor, Department of Civil and Environmental Engineering, Water Resources Research Center, University of Hawaii at Manoa, Holmes Hall # 383, Honolulu, Hawaii 96822, Tel: 808-956-7298; Fax: 808-956-5014; E-mail:rbabcock@hawaii.edu
Polymerase chain reaction denaturing gradient gel electrophoresis (PCR-DGGE) analysis and DNA sequencing was conducted to compare the bacterial communities that thrive in various wastewater treatment systems treating municipal wastewater. This study compares the microbial community structure from bench, pilot, and full-scale membrane bioreactors (MBRs), conventional activated sludge (CAS), and trickling filter solids-contact (TF/SC) systems. DGGE fingerprints obtained were examined by GelCompar II cluster analysis for community similarity. Bench and pilot-scale MBRs that treated the same wastewater had the highest similarity (73%) among all sites. Samples from a full-scale MBR treating a similar wastewater yielded a lower but still fairly high level of similarity (44%) to the bench and pilot-scale MBRs. Bacteria populations in CAS systems were more similar to each other (64%) than to the TF/SC systems (43%). The similarity between the MBR grouping and CAS plus TF/ SC grouping was in the significant low level (less than 5%) indicating that the dominant species in MBRs are very different than other forms of activated sludge treatment. Seventy-three percent of the excised DGGE bands were successfully identified by DNA sequencing which found four bacterial species that were present in more than one biological wastewater treatment system: uncultured Paracoccus sp. clone 3-3, uncultured Bacterium clone SB3-6, uncultured Clostridium sp., and uncultured Klebsiella sp. The results suggest that microbial community structure found in bench and pilot-scale MBRs may not be a good model for studying performance of full-scale MBRs due to non-identical operational conditions and internal hydrodynamic regime.
Wastewater treatment; Microbial community; PCR-DGGE; DNA sequencing
Membrane bioreactors (MBRs) have increased in popularity in recent years due to their ability to maintain high mixed liquor suspended solid (MLSS) concentrations, longer solids retention time (SRT),improved treatment performance and also offer high degree of operational flexibility [1,2]. MBRs are capable of producing an effluent quality similar to the quality obtained from the combination of secondary clarification and effluent microfiltration in a small foot print [3]. However, MBRs suffer unique challenges that limit their widespread application especially due to plant maintenance and operating costs as a consequence of membrane biofouling [4]. One method to control biofouling could involve physical, chemical, or biological means applied to control specific bacterial species known to cause fouling. In order to do so, the bacteria would need to be identified and then its habitat characteristics studied extensively to identify control strategies. It is also important to determine whether or not the bacterial communities are affected during scaling up of bench or pilot scale systems to full-scale systems such that control strategies developed/ tested at smaller scales that will also be valid for full-scale systems. It is also important to find out if the organisms in MBRs are different from those in other types of biological treatment processes such as conventional activated sludge (CAS) and trickling filter systems. Much of what is known or assumed concerning biological processes in MBRs has primarily come from investigations on CAS systems, regardless of the fact that significant differences in operating/environmental conditions exist between the two treatment processes. A few researchers such as Le-Clech et al. [4], Li et al. [5], Wu and Huang [6] have reported the effect of MLSS characteristics on membrane fouling. The link between microbial community structures to membrane biofouling has also been reported [7-9]. A shift in microbial community composition occurs with changed operating conditions and shift can lead to loss of ecosystem functions (nitrification, denitrification and biological phosphorus removal) [10].
In a study, Wan et al. [11] used denaturing gradient gel electrophoresis (DGGE) to analyze community structure and reported that the bacterial communities in pilot-scale MBRs fed with raw sewage were distinct from that in the CAS process. Miura et al. [12] analyzed bacterial community structure using polymerase chain reaction (PCR)-DGGE and fluorescence in-situ hybridization (FISH) techniques and reported that influent wastewater composition had a large impact on bacterial community structures. Changes in bacterial community structures in response to influent waste characteristics were also revealed by Stamper et al. [13].
There have been no molecular microbial diversity surveys on full-scale MBR systems for municipal wastewater treatment until recently by Wan et al. [11].It was shown that the bacterial communities in MBRs were constantly distinct from those in the parallel-running CAS bioreactors, reflecting the contrasting environmental and operational conditions in two treatment systems. However, not much research has been done to compare (presence and abundance) bacterial species in the parallelrunning bench, and pilot scale MBRs, and full scale trickling filter/ solids contact (TF/SC) processes treating municipal wastewater. Various researchers [14,15] have reported that bands on the same position in a DGGE profile may come from different source of bacteria and different bands may also come from the same source of bacteria [16,17]. Moreover, DGGE bands show the change in bacterial community structure over time. Despite such limitations, DGGE has been recognized as a rapid method for bacterial community analysis. Herein, a comparison of microbial community structure that thrives in MBRs with CAS, and TF/SC processes that treat municipal wastewater has been made by enumerating bacterial communities using PCR-DGGE technique.
Approximately one liter mixed liquor samples (aeration tank sewage sludge) were collected semi-monthly over a 6-month period from 4 different wastewater treatment plants (WWTPs). Samples were collected in autoclaved plastic bottles and preserved in ice immediately after collection and shipped to the lab and kept refrigerated till analyzed. All samples were analyzed at the University of Hawaii’s Water Resources Research Center lab. These four WWTPs (Honouliuli, Wahiawa, East Honolulu, and Schofield Barracks) receive different quality waste streams as the salinity and community size/characteristics (including residential, industrial, and commercial size) vary. Honouliuli WWTP is the second largest WWTP in the State of Hawaii in terms of design flow and is located in the Western part of the island of Oahu. This WWTP receives an average flow of 26 million gallons per day (MGD) of medium salinity (total dissolved solids (TDS) 795 mg/l) wastewater of mostly residential nature and uses TF/SC for biological treatment. The bench and pilot-scale MBRs were located at Honouliuli WWTP premises and ran in parallel with the same wastewater the TF/SC was receiving. Other WWTPs included in the study are the 4.5 MGD capacity East Honolulu WWTP treating high salinity (TDS 4400 mg/l) residential wastewater using CAS, the 2.5 MGD capacity Wahiawa WWTP treating low salinity (TDS 218 mg/l) residential quality wastewater using CAS, and the 4.2 MGD capacity Schofield Barracks WWTP treating low salinity (TDS ~254 mg/l) mixed residential/industrial wastewater using hollow-fiber type MBR.
Total genomic DNA was purified directly from mixed liquor using Fast DNA Spin Kit (MP Biomedicals, Solon, OH, USA) followed by amplification into fragments by PCR. Using the v3 region of 16S rRNA as template, forward Primer 338f(5’-ACT-CCT-ACG-GGA-GGC-AGCAG-3’)as reported by Lane [18] and reverse primer 518r (5’-ATT-ACCGCG-GCT-GCT-GG-3’) as suggested by Muyzer et al. [14] and Øvreås et al. [19]were used as universal primers for PCR amplification. The 50 µl PCR mixtures contained: 2.5 µl of each primer, 1 µl of 10 mM PCR Nucleotide Mix, 10 µl of 5x colorless Go Taq Flexi reaction buffer, 5 µl of 25 mM MgCl2 solution, 0.25 µl of 5 u/µl Go Taq DNA polymerase, 2 µl of BSA, 26 µl of nuclease-free water, and 0.75 µl of extracted DNA. PCR amplification was performed in an iCycler (Bio-Rad Laboratories, Hercules, CA, USA). The 16S rRNA gene amplification had an initial denaturation at 94°C for 5 min; 35 cycles of denaturation (45 sec at 94°C), annealing (45 sec at 55°C) and extension (45 sec at 72°C), and a final extension at 72°C for 10 min. The PCR-amplified DNA samples were examined by horizontal electrophoresis in 1.5% agarose with 2 µl aliquots of PCR products as described [14].
After PCR amplification, DNA samples were loaded into 8% wt/ vol polyacrylamide gels (37.5:1, acrylamide/bisacrylamide) in 1 × TAE buffer (20 mM Tris, 10 mM acetate, 0.5 mM EDTA, pH = 7.4). The gel ran under 130V at 60°C for 210 min in Bio-Rad D Code system (BioRad, Hercules, CA). The gel was then stained in ethidium bromide (1.0 mg/l) for 10 min, rinsed in 1 × TAE buffer for 10 min, and visualized with a UV trans illuminator (Bio-Rad Gel Doc 2000, Bio-Rad, Hercules, CA).Specific DGGE bands were then manually excised from the gel for DNA sequencing. Sequencing methods are described in Huang et al. [20]. Several methods were used to improve the low DNA sequencing results obtained (less than 67% identified initially). It was reported that betaine improves the amplification of genes by reducing the formation of secondary structure caused by GC-rich regions [21]. The addition of 5 M betaine into DNA samples before sequencing increased the results by 4%. Increased annealing temperature from 50°C to 60°C and extension temperature from 60°C to 80°C resulted in no sequence results. However, sequence results increased by 3% after increasing temperature from 50°C to 58°C and extension temperature from 60°C to 70°C during sequencing according to the primer P518r (64.7% GC content) (Tm = 4°C x (number of G’s and C’s in the primer) + 2°C x (number of A’s and T’s in the primer)). The simultaneous use of 5 M betaine and increase in annealing temperature to 58°C and extension temperature to 70°C during sequencing increased the results by 6%. Although sequencing conditions improved, 27% of DNA sequencing results remained unidentifiable.
After the DGGE study, 4 representative samples from each wastewater treatment system were chosen for comparative analyses. GelCompar II cluster analysis was used to compare the structure and similarity between bacterial communities from different WWTPs (Applied Maths, Austin, TX, USA). Global similarity is calculated by merging characters or by averaging experiment-related similarities using a Pearson correlation, unweighted pair grouping (UPGMA). It is noted that DGGE is thought to be best suited to identifying the major populations and not the minor, but possibly very important, populations in a diverse bacterial community. Individual DGGE bands were excised from the DGGE gels and then subjected to DNA sequencing and evaluated using the online NCBI gene bank database to identify individual bacterial species.
For community diversity analysis, it was assumed that each band corresponded to a unique species, with band density corresponding to species abundance. Each band was considered to come from one source of bacteria during the bacterial community diversity study. Moreover, each DGGE profile pattern can display one or more different sample specific dominant species. The differences in observed sample specific dominant species under each operating condition were considered caused by different operational/environmental environments that the microbial communities were exposed to leading to different physiological growth conditions.
Figure 1 below (1a: left part and 1b: right part) shows DGGE analysis comparison among bench-scale, pilot-scale and full-scale MBR samples, and East Honolulu CAS, Wahiawa CAS and Honouliuli TF/SC processes, respectively.
As depicted in Figure 1, the DGGE fingerprint for the bacteria community in the bench-scale MBR contained 10 re-occurring bands; 5 of them permanent bacteria species (bright band representing a dominant microbial species and consistently present over 6 month study period). Among those five species, band B2, identified as uncultured Bacterium clone Pia-s-66, and showed a relatively greater dominance in terms of band brightness (Table 1). Nine of the 10 species were identified via sequencing including uncultured Paracoccus sp. Clone 3-3 (band B7). Uncultured Paracoccus sp. Clone 3-3 was also found in samples from full scale Schofield MBR and TF/SC process at Honouliuli. Results were significantly different between the bench-scale and the pilot-scale MBRs from same manufacturer and processing same waste stream with 6 of 9 major species identified. Only band P2 identified as uncultured Eubacteriumclone F10.18 16S showed stability throughout the entire period of tracking representing only one permanent bacterial species in the pilot system.
Figure 1: DGGE fingerprints of PCR amplified 16S rRNA fragments extracted from samples of mixed liquor from bench, pilot, and full-scale (Schofield) MBRs; and from East Honolulu CAS, Wahiawa CAS, and Honouliuli TF/SC.
In the initial setup of the reactor, Flavobacterium sp. AKB-2008-JO36 (band P3) demonstrated the greatest abundance (displaying the brightest band quality) but faded over time as system reacclimatization occurs. The PCR-DGGE banding patterns of the bacteria communities in the fullscale MBR at Schofield (different waste stream from bench and pilot) and TF/SC at Honouliuli (same waste stream as bench and pilot MBRs) were each very constant during 6 months of tracking. The DGGE fingerprint for Schofield MBR had 16 bands that appeared in all samples with 13 identified via sequencing. The TF/SC plant at Honouliuli also had 16 bands in all samples and all 16 were identified (Figure 1a, bottom part). CAS plants were somewhat less stable over time with the high-salinity East Honolulu plant showing less stability of bacteria speciation. Four bacteria species identified in samples from Honouliuli (TF/SC) WWTP were also found in the samples from the other treatment plants (Table 1).
While the 16S rRNA fingerprints for MBRs were dominated by a limited number of bands, the CAS fingerprints displayed complex banding patterns and thus indicated a diverse bacterial community. In the East Honolulu CAS samples, there are 15 detectable bands with 10 of them successfully identified via sequencing (Figure 1b). Similarly, there are 14 detectable bands in the Wahiawa CAS samples but only 7 of them were identified to the closest phylogenetic affiliations. All samples had high similarity to each other indicating that the biological system stressors were relatively constant. Each bioreactor showed a diverse bacteria community and different dominating bacteria species as indicated by the different banding patterns and the brightness of each band, respectively.
The Schofield Barracks full-scale MBR samples have more bands than the bench-scale and pilot-scale samples. Also, the bench and pilot-scale MBR systems have greater continuity in the appearance of the bands. Cluster analysis (Figure 2) revealed that bench-scale MBR and pilot-scale MBR have 72% similarity, the highest similarity among all sample sites tested. However, the results indicate that the bench-scale MBRs do not harbor the same dominant population as the pilot scale MBRs. These systems were seeded with the same sludge, utilized the same membranes (same manufacturer, pore size and configuration), and treated the same waste stream side-by-side at the same SRT. However, the bench scale unit had different physical dimensions resulting in different hydrodynamics (larger hydraulic retention time, different recycle rates, and different aeration/mixing regimes) indicating the role played by these parameters in selecting microbial population. Schofield full-scale MBR samples have a lower level of similarity (44%) with bench-scale and pilot-scale MBR samples. This could be partly explained by the different waste stream treated by the Schofield full-scale MBR (low salinity, mixed residential/ industrial) and the bench and pilot-scale MBRs (medium salinity, residential) and also the non-identical reactor internal configuration and operation as compared to the small-scale units. DGGE analysis of PCRamplified 16S rRNA fragments from East Honolulu CAS, Wahiawa CAS, and TF/SC at Honouliuli revealed significant differences amongst the samples (Figure 2).

Table 1: Sequencing result showing Genbank accession number, closest phylogenetic affiliation, and % sequence similarity
Cluster analysis showed that East Honolulu CAS and Wahiawa CAS have 64% similarity while Honouliuli TF/SC samples have a lower level of similarity (43%) with the CAS facility samples. Figure 2 also shows that the similarity between the CAS and TF/SC group and the MBR group is in the significant low level (less than 5%). Although the bench-scale, pilotscale, and TF/SC at Honouliuli all treated the same waste stream, there are major dissimilarities in the dominant microbial communities. Of the two environments are likely due to the complete rejection of biomass by the membrane and the contrasting operating conditions applied (e.g. sludge age, F/M ratio) in the MBRs. Clearly, the MBRs select for a dominant bacteria population that is different from that of CAS and TF/SC systems.
Generally, both operational and environmental conditions should affect bacterial community composition and which organisms are dominant. In Hawaii there is essentially no variation in wastewater temperature on either temporal or special bases so it is not considered a selection pressure. Municipal wastewater has a complex composition that is quite variable over time and thus microbial community has to be diverse and adaptable to environmental conditions to ensure consistent treatment results. The MBR systems have longer SRTs (approximately 30 days versus 10-15 days for CAS and TF/SC) and higher MLSS (approximately 10,000 mg/L versus 2000 mg/L for CAS and 3,500 for TF/SC) that causes a low F/M which allows endogenous decay of biomass and formation of more recalcitrant DOM, the larger size fractions of which are retained in the aeration tank by the membrane. The formation and retention of soluble microbial products (SMP) and extracellular polymeric substances (EPS) may also favor growth of different organisms that scavenge these products in the MBR food web. Shorter hydraulic retention time, greater shear forces (greater aeration rates using coarse bubble aeration), and retention of all biomass by membranes causes different selection pressures as described by Wan et al [11]. All of this indicates that the physiological state of the biomass is different in MBRs as compared to CAS systems. The TF/SC mixed liquor is only exposed to the residual organics coming off of the TF biotowers and thus it also operates in a low F/M condition (possibly even lower than the MBRs), however, the environmental conditions are much more similar to CAS than MBRs.
Influent wastewater characteristics can account for microbial diversity variations and that surely played some role in the differences observed between the CAS systems with Wahiawa CAS (low-salinity) and East Honolulu CAS (high-salinity) because both systems treat residential-type municipal wastewater with similar organic and nutrient compositions. The wastewater at Honouliuli TF/SC is considered medium salinity, mostly residential municipal wastewater. However it is a larger regional plant with a large collection area such that the influent is aged (septic) which could account for some of the differences in community diversity. However, the proposed major factor is the difference in the operational/ environmental conditions between TF/SC and CAS with the TF/SC mixed liquor only exposed to the residual organics coming off of the TF biotowers.
DNA sequencing also revealed that four bacterial species were found in more than one wastewater treatment system: (a) uncultured Paracoccus sp. clone 3-3 was found in bench-scale and Schofield full-scale MBRs, and Honouliuli TF/SC. This is a genus of Rhodobacteraceae which are gram- negative heterotrophs common in aquatic environments and wastewater, (b) uncultured Bacterium clone SB3-6 was found in pilot-scale MBR and Honouliuli TF/SC, (c) uncultured Clostridium sp. found in East Honolulu CAS and Honouliuli TF/SC. These bacteria are gram-positive anaerobic bacteria that are common in mixed liquor and some of the 100 species are important pathogens, and (d) uncultured Klebsiella sp. was found in Wahiawa CAS and Honouliuli TF/SC. These bacteria are gram-negative facultative members of the Enterobacteriaceae family found everywhere in nature. These four organisms do not necessarily play a dominant role in these systems, but the fact that they were identified at multiple facilities possibly indicates their common nature in terms of substrate preference/ availability as well as possible physiological competitive advantage.

Figure 2: Comparison of bacteria communities obtained from East Honolulu CAS, Wahiawa CAS, Honouliuli TF/SC, Bench-and Pilot-Scale MBRs at Honouliuli, and Schofield Full-Scale MBR analyzed by GelCompar II. Curve based: Pearson correlation; Method: Unweighted pair-grouping (UPGMA).
Seventy-three percent of the DGGE bands were successfully sequenced to the closest phylogenetic affiliation and several of the same bacteria species were found in the mixed liquor form multiple wastewater treatment plants. Cluster analysis of DGGE bands showed that cultures from bench-scale and pilot-scale MBRs treating the same wastewater have a 72% similarity with each other but full-scale MBRs had a lower level of similarity (44%) with the smaller-scale MBRs. The East Honolulu CAS and Wahiawa CAS plant cultures showed 64% similarity (residentialonly municipal waste but quite different salinity),while there was a lower similarity (43%) between the CASs and the Honouliuli TF/SC that is thought to be due to both different waste characteristics and operational/ environmental regimes).The similarity between the MBR group and the non-MBR group is in the significant low level (less than 5%) due to very different operational regimes resulting in different physiological states of the cultures even if treating wastewater with the same or similar characteristics. Because microbial populations appear to differ amongst bench, pilot and full-scale MBRs and CAS and TF/SC processes, care has to be taken in extrapolation of data collected from bench and pilotscale studies to expected effects in full scale MBRs. Additional research is needed to explore the influence of operating conditions on the diversity and abundance of microbial communities in MBRs since most of the studies have focused on the performance of the MBRs and membrane fouling without considering the bacterial communities involved.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation: Xu-Sadri H, Lamichhane KM, Babcock R (2015) Analysis and Comparison of the Bacterial Community in Membrane Bioreactors and Other Treatment Systems. Int J Water Wastewater Treat 2(1): doi http://dx.doi.org/10.16966/2381-5299.112
Copyright: © 2015 Xu-Sadri H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access