Introduction
Upon viral infection host pathogen recognition receptors, including the Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), detect the presence of foreign motifs referred to as pathogen-associated molecular patterns (PAMPs) and activate a signaling pathway that ultimately leads to the induction and expression of the type 1 interferons (IFN). This newly produced IFN establishes an antiviral state in surrounding cells that prevents virus replication. Therefore, induction of IFN gene expression and the activation of subsequent IFN signaling pathways is crucial to the ability of a host cell to mount an innate immune response [1]. To counteract these powerful antiviral responses many viruses have evolved elegant, and often multi-pronged, mechanisms by which they evade the innate immune response [2]. There has been a tremendous amount of research done to understand how different viruses block induction of the IFN gene by either preventing recognition by RLRs or suppressing the signaling pathways they activate. One well-studied member of the RLR family is the retinoic acid–inducible gene-1 (RIG-I). This cytoplasmic receptor primarily detects 5′ppp-RNA molecules with short secondary motifs of dsRNA or ssRNA [3,4]. In contrast, another cytoplasmic RLR referred to as MDA5 recognizes longer dsRNA motifs so that each RLR recognizes different viruses based on their respective PAMPs [5]. Following binding of viral RNAs, RIG-I and MDA5 interact with the mitochondrial membrane bound adaptor molecule MAVS (mitochondrial antiviral signaling protein, also referred to as IPS-1, VISA, or CARDIF), which activates two kinase complexes. The IκBKinaseɛ/ TANK Binding Kinase 1 (IKKɛ/TBK1) phosphorylate the transcription factors, interferon regulatory factors (IRF), IRF3 and IRF7, which then form homodimers or heterodimers, enter the nucleus and initiate transcription of IFNα/β. For clarity, it is worth mentioning that the type I interferons include a subgroup of interferon proteins that include IFNα/β. While IRF3 is constitutively expressed in most cells, IRF7 is an interferon stimulated gene (ISG) that is typically expressed at low levels but can be induced several-fold in response to IFN signaling. Therefore, it is thought that IRF3 mediates transcription of the majority of early IFN expression. The IKKα/IKKβ/IKKγ kinase complex phosphorylates IκBα, targeting this repressor protein of nuclear factor kappa B (NF-κB) for degradation. Following secretion outside of the initially infected cell, the IFN protein is recognized by target cells and initiates their IFN signaling pathways [1,6]. Ultimately this leads to the expression or upregulation of hundreds of ISGs, including IFN, pro-apoptotic factors, and cytokines which establish an antiviral state in surrounding cells [1,7].
This review will focus on how select RNA viruses evade the innate immune response. Specifically, we will focus on how the top eight emerging viruses, as identified by the World Health Organization [8], suppress RIGI-mediated induction of the IFN antiviral response as shown in Figure 1. In order to provide perspective, we also include information about how vesicular stomatitis virus (VSV), a well-studied non-human pathogen, evades the host immune response. VSV serves as a model for how nonhuman pathogenic RNA viruses act in manners both similar to and different from the other emerging viruses. Taken together, the diversity of mechanisms employed by these pathogens to circumvent host defenses is remarkable. The similarities as well as the differences are striking.
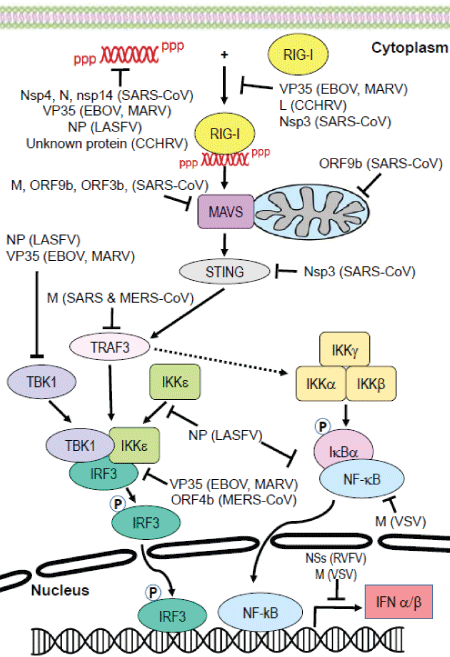
Figure 1: Targeting of the RIG-I signaling pathway by emerging virusesUpon activation by cytoplasmic RNA, RIG-I is activated and interacts with MAVS. This initiates downstream signaling events that activate IRF3 and NF-κB, and ultimately results in induction of the IFNα/β gene. Many components in this pathway are inhibited by viral proteins, thereby suppressing the IFN response and enabling viral replication to occur. Viruses depicted above include Rift Valley fever virus (RVFV), Crimean-Congo hemorrhagic fever virus (CCHFV), ebolavirus (EBOV), Marburgh virus (MARV), Lassa fever virus (LASFV), Nipah virus (NiV), severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome virus coronavirus (MERS-CoV) and vesicular stomatitis virus (VSV).
Rift Valley Fever Virus and Crimean-Congo Hemorrhagic Fever Virus
Members of the Bunyaviridae family that are listed in the 2016 WHO list of emerging viruses include the zoonotic arthropod-borne Rift Valley fever virus (RVFV) and the Crimean-Congo fever virus (CCHFV). Both of these viruses carry a tripartite negative sense RNA genome [9] and can cause severe disease in humans, including fulminating hemorrhagic fever [10,11].There are currently no prophylactic or therapeutic treatments available for these viruses [9]. The pathogenicity of these viruses is largely attributed to the ability of the multifunctional nonstructural protein NSs to inhibit global host cell transcription and to antagonize the IFN system [9,12-14].
Although RIG-I is activated upon recognition of RVFV RNA [15], IFN production is delayed in RVFV-infected animal models [13]. Several studies have demonstrated that the NSs protein utilizes several mechanisms to block IFN-β gene expression during early RVFV infection [13,16,17]. NSs was found to directly target IFN- β gene expression through its interaction with the cellular repressor protein Sin3A-associated protein 30 (SAP30), a subunit of the Sin3A/nuclear receptor co-repressor (NCoR)/histone deacetylase repressor complex. NSs simultaneously interacts with YY1, a transcription factor that regulates IFN-β gene expression [18]. YY1 directs the SAP30-NSs-YY1 complex to the IFN- β promoter site to form a multi protein repression complex on the promoter, which inhibits induction of the IFN-β gene [17]. RVFV NSs also indirectly down regulates IFN-β gene expression by shutting-off global host gene transcription by sequestering the p44 and XPD subunits of the TFIIH basal transcription factor [19]. NSs also inhibits host transcription by promoting the degradation of the TFIIH p62 subunit [20].
Similarly, IFN production and secretion is delayed during CCHFV infection [21,22]. A virally encoded protease processes the CCHFV genome to include a 5′ monophosphate (5′p) end [23], rather than the 5′ppp and 5′pp ends strongly recognized by RIG-I [24]. Therefore it was proposed that due to this modification CCHFV RNA is not sensed by RIG-I [23,25]. However, recently it was established that RIG-I does mediate an IFN response to CCHFV [26]. In fact, immuno stimulatory RNA (isRNA) was isolated from infected cells as well as from virion preparations, and RIG-I co-immuno precipitation resulted in the isolation of CCHFV isRNA from infected cell lysates. These findings indicate that RIG-I signaling is critical to the activation of an antiviral response to CCHFV infection [26].
While the CCHFV protein that antagonizes RIG-I-dependent IFN production has not yet been identified, the viral L protein has been suggested as a potential candidate. In addition to functioning as the viral RNA dependent-RNA polymerase, the CCHFV L protein is a cysteine protease that contains a viral homologue to the ovarian tumor protease domain (OTU) [27], which allows the removal of conjugated poly-ubiquitin (Ub) and interferon-induced Ub-like protein (ISG15) from target proteins [28,29]. Viral proteases which contain this domain evade ubiquitin- and ISG15-dependent innate immune responses [27,30], therefore it is possible that the CCHFV OTU directly antagonizes the innate immune response. More research must be done to determine if the CCHFV OTU blocks RIG-I signaling and to identify which proteins in the RIG-I pathway are targeted for OTU-dependent de-conjugation of Ub and ISG15.
Ebola and Marburg Viruses
Ebolavirus (EBOV) and Marburg virus (MARV) are members of the Filoviridae family that infect primates. They can cause hemorrhagic fever and are among the most virulent pathogens known, with case fatality rates reaching 90% during some outbreaks [31]. Mortality is swift and follows the shock and subsequent multi-organ failure that results from hemorrhagic complications [32]. This virulence is attributed to virally encoded proteins that antagonize the ability of the host to mount an effective innate immune response, leading to uncontrolled virus replication. It has been demonstrated that EBOV VP24 and the MARV VP40 inhibit the IFN signaling pathway [33,34]. As this occurs during the later phase of the IFN response it will not be discussed further herein.
In addition to its function as a polymerase cofactor and its role in viral assembly, the EBOV VP35 (eVP35) and MARV VP35 (mVP35) suppress innate immunity by targeting multiple steps in the RIG-Idependent induction of IFN gene expression [35,36]. Both eVP35 and mVP35 bind dsRNA [37] through a basic amino acid motif located in the highly conserved C-terminal IFN-inhibitory domain (IID). This binding sequesters the dsRNA from RIG-I surveillance and therefore prevents IFN production. The IID domain interacts with dsRNA in a sequencespecific manner and was demonstrated to be essential for VP35-mediated inhibition of IFN production [38-41]. By binding to viral dsRNA, eVP35 inhibited activation of the IFN-β promoter normally induced by overexpression of RIG-I, MAVS, IKKε and TBK1 [37]. Mutation of dsRNA-binding residues led to a decrease in dsRNA binding [37,42].
Comparison of the crystal structures of eVP35 and mVP35 IIDs bound to dsRNA revealed that eVP35 interacts with both the phosphodiester backbone and caps the ends of dsRNA [40,43], while mVP35 was found to interact with the dsRNA backbone only [41]. Edwards and coworkers established that eVP35 was able to more strongly inhibit RLR signaling than mVP35. This correlated with induction of a more robust IFN response in MARV-infected cells as compared to EBOV-infected cells. These functional differences between eVP35 and mVP35 mapped to IID. Therefore the binding mode of both viral VP35s with dsRNA plays a significant role in the magnitude of the IFN response in filoviral-infected cells [44].
While VP35 has been shown to bind synthetic dsRNA molecules introduced in vitro [45], direct evidence that VP35 binds isRNA to limit RIG-I activation was lacking. Utilizing a Sendai virus (SeV) infection model and deep sequencing of purified eVP35-bound RNAs, Dilley and coworkers demonstrated that the SeV defective interfering (DI) RNA, a known activator of RIG-I, is the is RNA bound by eVP35 proteins in infected cells. Mutation of basic residues in the IID domain that were required for dsRNA binding and inhibition of IFN destroyed the ability of eVP35 to bind the SeV DI RNA. In addition, select host RNAs were preferentially bound by wild type eVP35 in cell culture. These findings support the contention that VP35 binds viral isRNA to block the RIG-I pathway and thereby evade the IFN response [45]. VP35 also inhibits IFN production by targeting the RLR pathways in a dsRNA bindingindependent manner by interacting with key components of the RIG-I pathway.
The IID was critical for the ability of eVP35 and mVP35 to block IRF3 phosphorylation and activation by over expression of IKKɛ and TBK1, the kinases that activate this transcription factor [38,46]. In contrast, these viral proteins did not inhibit IFN-β promoter activation induced by expression of a constitutively active form of IRF3 [41,47]. Interestingly, eVP35 was found to target and bind to the N-terminal domain of both IKKɛ and TBK1 and was subsequently phosphorylated by these kinases. Overexpression of eVP35 and its interaction with IKKɛ and TBK1, sequesters them and impairs their normal interactions with IRF3, IRF7, and MAVS, and decreases the kinase activity in cells transfected with IKKɛ [47]. Taken together, these findings indicate that VP35 can act as a decoy substrate for the TBK1-IKKε complex, thereby impairing IRF3 phosphorylation through its normal interaction with TBK1 and IKKɛ [37,47].
Expression of wild-type eVP35 also interferes with the ability of RIG-I to interact with PACT, a cellular dsRNA binding protein that is an essential coactivator of RIG-I [48]. Mutations in the eVP35 IID domain prevented eVP35-PACT binding and limited the ability of eVP35 to inhibit PACTmediated activation of RIG-I. Cells in which PACT had been knocked down were defective for IFN induction and were insensitive to eVP35 activity [49].
It has been shown that TLR and RIG-I signaling covalently conjugates SUMO molecules to both IRF3 and IRF7 and this modification was correlated with reduced IFN transcription [50]. In addition, physical interaction of eVP35 with IRF3 and IRF7 led to their sumoylation. This modification inhibited the transcriptional activity of these IRFs and the downstream expression from the IFN-β promoter [51].
Lassa Fever Virus
Like other members of the Arenaviridae family, Lassa fever virus (LASFV) is an enveloped negative-sense RNA virus that carries a bisegmented genome [52]. LASFV is endemic in several West African countries where there are between 300,000-500,000 cases annually. This virus can cause fatal hemorrhagic fever in humans, resulting in approximately 5,000 deaths per year [53-56]. The pathogenesis of LASFV is associated with the ability of this virus to specifically target dendritic and endothelial cells [57,58]. In addition, LASFV is able to suppress the induction of host IFNs.
While the 5′-ppp dsRNA associated with the LASFV genome activates the RIG-I pathway [23], the virally encoded protein, NP, was identified as an IFN antagonist [59-61]. By inhibiting IRF3 phosphorylation, the multifunctional NP suppresses IFN induction [60,62]. This function of the LASFV NP is dependent on its intrinsic 3′–5′ exoribonuclease (ExoN) activity, which digests free dsRNA and thereby prevents RIG-I recognition of that non-cellular nucleic acid [63,64]. Mutations in the exoribonuclease active site dramatically reduced this activity and abrogated the ability of the LASFV NP to inhibit viral- or synthetic polyI:C-induced activation of the IFNα/β promoter in vitro [63-65]. Importantly, residues essential for NP-mediated IFN inhibition are highly conserved among all arenaviruses, indicating that this function too is conserved across all members of this viral family [63,65,66]. A robust, RIG-I dependent, innate immune response was activated in cells infected with a recombinant LASFV in which the ExoN function was abolished. These results correlate with earlier in vitro studies and underscore the essential role of the NP exonuclease activity in suppression of innate immunity during LASFV infection [67].
This same region within the NP protein was found to antagonize induction of IFN gene expression by inhibiting the nuclear translocation and transcriptional activity of NF-κB [68] and by blocking the autocatalytic activity of IKKɛ. By binding to the kinase domain of IKKɛ, NP inhibited the ability of the kinase to phosphorylate, and therefore activate IRF3. This NP-IKKɛ interaction also prevented IKKɛ from interacting with MAVS, thereby blocking the RIG-I pathway [69]. Interestingly, mutation of the same NP residues that are critical for its 3′–5′ exoribonuclease activity perturbed the interaction of NP with IKKɛ[69].
Nipah Virus
Nipah virus (NiV), also identified as an emerging virus, is a lethal pathogen that causes death in up to 70% of infected humans [70]. This virus infects both bats and humans but most likely originated in the former [71]. While other paramyxoviruses, such as Hendra virus, also use bats as a natural reservoir they do not all infect both bats and humans [72]. In fact, Hendra virus and NiV may be the only two and they are both lethal in humans [73]. One study suggested that bat to human transmission, and therefore the risk of human infection, is increased in those individuals who drink tree sap [71]. Other studies have elucidated the mechanisms employed by NiV to evade host innate immune responses.
When Pteropus vampyrus bat kidney (PVK) cells are infected with the related avian Newcastle disease virus (NDV), Glennon and coworkers observed an increase in expression of the genes encoding IFN, the GMCSF and IL-2 inhibitory factor I (GIF-I) and MDA5, among others [74]. In contrast, when those same cells are infected with NiV these genes are not upregulated, suggesting that NiV, perhaps uniquely, antagonizes expression of these host genes to facilitate viral replication. Suppression of IFN expression is most likely achieved by the viral accessory proteins V, W and C [75]. Similar responses involving the viral C protein have been observed in cells infected with measles virus [76]. In that system the suppression is most likely achieved by a combined mechanism that includes suppression of Janus Kinase 1 (jak1) phosphorylation and associated effects of the viral C protein [77]. The diversity observed in the ways different paramyxoviruses suppress host antiviral responses suggests that not only are their biological differences interesting but potential therapeutic approaches must be targeted to specific viral pathogens.
Severe Acute Respiratory Syndrome and Middle East Respiratory Syndrome Viruses
The Severe Acute Respiratory Syndrome Corona Virus (SARS-CoV) was first identified in 2002 in China as the causative agent in those affected individuals presenting with respiratory complications after exposure to a single health care worker [78]. Within eleven weeks of the first incidence in neighboring Hong Kong, the virus had spread to at least 27 countries or distinct political entities with nearly one fourth of the reported cases occurring among health care workers [79]. A wide range of fatality rates have been reported and not surprisingly they vary by location and they decrease over time [80,81]. The Middle East Respiratory Syndrome coronavirus (MERS-CoV) is another highly pathogenic member of this family. This lethal virus appears to be carried by Dromedary camels and is transmitted directly from them to humans [82]. When discovered in 2012 the virus displayed a nearly 37% mortality rate [82].
Patients with severe SARS disease displayed dysregulated IFN, ISGs and cytokine responses [83]. Similarly, MERS-CoV-infected cells exhibited reduced IFN and cytokine expression, blocked IRF3-mediated induction of the IFN response and upregulation of RIG-I, IRFs and other genes associated with innate immunity [84-86]. Taken together, these findings strongly suggest that the extreme virulence of SARS-CoV and MERSCoV is related to their ability to evade the host innate immune response.
SARS-CoV may hide its dsRNA from detection by RIG-I by replicating in “inner vesicles” within the lumen of a virus-induced reticulo vesicular network of modified endoplasmic reticulum (ER) membranes. The viral replicase (composed of the nsp3, nsp5, and nsp8 proteins) as well as the viral genomic RNA co-localize to these double membrane vesicles (DMVs), providing evidence that SARS-CoV replicates in this membrane network. The interior of these DMVs label for SARS-CoV dsRNA, therefore this virus forms DMVs to coordinate its replication and also hide replicating RNA from RLRs. The nsp4 viral replication protein appears to direct this membrane rearrangement, as its mutation alters assembly of these DMVs [87]. Interestingly, a similar phenomenon was observed in MERS-CoV-infected cells [88], indicating that at least two coronaviruses hide their dsRNA inside DMVs, avoiding detection by the host [89]. The SARS-CoV nucleocapsid (N) protein may suppress IFN production via a similar mechanism. Studies indicate that the N protein suppresses IFN signaling by targeting an early step in the pathway [90,91] and binds to dsRNA [51,92]. Therefore the N protein likely plays a key role in blocking the innate immune response [91] by shielding dsRNA from recognition by RIG-I. The SARS nsp14 protein contains a 3’-5’ exoribonuclease domain, therefore this protein may function to limit the IFN response by degrading viral dsRNA replication intermediates. Indirect support for this notion comes from studies of the LASFV encoded NP which contains a similar exonuclease domain. Mutation of critical residues within this domain abrogated the ability of LASFV NP to inhibit induction of the IFNα/β promoter [63-65]. While it is conceivable that the SARS-CoV nsp14 protein suppresses the IFN response by degrading dsRNA, further work is required to determine if this is indeed the case. Nevertheless, it is interesting that similar approaches are employed by viruses from different families. In this case an arenavirus and a coronavirus.
Several other proteins encoded by SARS-CoV antagonize the RIG-I signaling pathway. For example, the ORF9b protein suppresses innate immunity by targeting mitochondria and MAVS/TRAF3/TRAF6. Expression of ORF9b altered the mitochondrial morphology and subcellular localization of MAVS. The presence of ORF9b also led to the ubiquitination and degradation of MAVS, accompanied by a loss of TRAF3 and TRAF6, two key components of the RIG-I signaling pathway [93]. The SARS-CoV ORF3b and ORF6 proteins limit RLR-mediated induction of IFN. ORF3 localized to the mitochondrial outer membrane and may therefore inhibit MAVS at the mitochondria or at a point downstream of MAVS [90,94]. In contrast, ORF6 localized primarily to the ER and Golgi apparatus and may disrupt the ER/Golgi transport necessary for the IFN response [90]. The SARS-CoV M protein inhibits induction of IFN by binding to TRAF3 and impeding the formation of a TRAF- ·TANK·TBK1/IKKɛ complex, thereby inhibiting TBK1/IKKɛ-dependent activation of IRF3 and IRF7 [95]. Finally, the papain–like protease (PLP) domain of the SARS-CoV nsp3 protein interacts with STING and disrupts the dimerization and activation of this adaptor molecule. Inactive STING is unable to recruit MAVS to the TBK1-IKKɛ complex, therefore these kinases do not phosphorylate IRF3 and IFN gene expression is not induced. The PLP domain of nsp3 also disrupts NF-κB signaling, possibly by a similar mechanism [96] and it expresses a deubiquitinating activity that removes Ub from key components of the pathway, including RIG-I, STING, TBK1 and IRF3 [96,97].
Expression of the MERS-CoV ORF4b antagonizes the host IFNα/β expression that is normally upregulated in response to viral infection [98]. The accessory protein encoded by ORF4b, termed p4b, acts in both the cytoplasm and the nucleus [99]. Interestingly, Yang and coworkers demonstrated that in the cytoplasm p4b binds to TBK1 and IKKɛ, thereby suppressing molecular interactions between MAVS and IKKɛ, while inhibiting the phosphorylation of IRF3 [98]. When in the nucleus, the same protein inhibits the IRF3 and IRF7 induced expression of IFN-β. However, ablation of the protein’s nuclear localization signal eliminated its ability to inhibit IFN- β expression but not the IFN- β expression induced by RIG-I, TBK-1, MAVS, MDA5 and IKKɛ. This suggests that p4b employs multiple approaches to inhibit IFN- β in both the cytoplasm and the nucleus, no doubt contributing to the observed viral pathogenicity. Interestingly, the MERS-CoV M protein is able to interact with TRAF3 which hampers the TRAF3-TBK1 interaction and therefore leads to a decrease in IRF3 activation. The N-terminal transmembrane domain of the MERS-CoV M protein is sufficient for interaction with TRAF3 [100], which is similar to what has been shown for the SARS-CoV M protein [101].
Vesicular Stomatitis Virus
While not on the WHO list of emerging viruses, VSV is a well-studied member of the Rhabdoviridae with a host range that includes insects, cattle, horses and pigs, and it serves as an excellent model system to study the interplay between viruses and the IFN responses of their hosts. The absence of IFN induction in wild type virus infected cells is thought to result from the presence of one or more virally encoded IFN suppressors that presumably are defective in IFN-inducing viruses [102]. One of these suppressors is the matrix (M) protein which is crucial for many of the cytotoxic effects associated with VSV infection, including the downregulation of global host gene expression [39,78,103] and inhibition of the nuclear-cytoplasmic transport of host mRNAs [9,11,104]. The M protein has been shown to inhibit host transcription [39,103] and suppress IFN-β gene expression in the absence of other viral components [78]. Therefore, several researchers have proposed that VSV evades the IFN response by an M-mediated “shut-off ” of host gene expression. In support of this hypothesis there is a strong correlation between the virus’s ability to inhibit host gene expression and its ability to suppress IFN expression.
Wild type VSV rapidly inhibits host RNA and protein synthesis and is a poor inducer, or non-inducer, of IFN [22]. In contrast, the VSV mutant strain T1026R1 [103], which contains a single amino acid mutation at position 51 (M51R) of the M protein [105], is delayed in its ability to inhibit host RNA and protein synthesis [106] and is an excellent inducer of IFN [21,30]. A recent study indicates that the M protein either in the context of viral infection or when expressed alone is able to block viralmediated activation of NF-κB by targeting a step in the canonical NF-κB pathway, and the M51R mutation abrogates this function [107]. These results imply that the VSV M protein encodes two suppressors of IFN gene expression; the well-described ability to inhibit host gene expression as well as the ability to suppress induction of the IFN-β promoter by specifically interfering with the NF-κB pathway. This is similar to the molecular strategies used by the RVFV NSs protein, which inhibits IFN gene expression indirectly by inhibiting global host transcription and directly by forming a multiprotein repression complex on the IFN gene promoter.
Conclusion and Recommendation
Many of the emerging viruses discussed herein are lethal to humans. While VSV is not lethal, it serves as a well-studied model of virus infection and host immune detection and has revealed mechanisms of host innate immune evasion that are seen in other viruses. Interestingly, even within families of viruses the approaches used by the individual viruses to thwart host innate immune surveillance vary. In contrast, some approaches are shared among viruses of different families. Taken together, this tangled story of host immune evasion by disparate RNA viruses makes the prospect of using a single therapeutic approach impossible. Therefore it is imperative that we better understand the specific interactions between virally-encoded proteins and those of their hosts in order to develop lifesaving therapies.