Introduction
There have been about 20 Ebola outbreaks since 1976, when the virus was first described [1], but until recently, most of the outbreaks were confined to isolated villages in Central Africa, with about 300 casualties in each episode. But by early 2014, ZEBOV emerged again and quickly spread to Liberia, Guinea, and Sierra Leone in West Africa and the number of casualties increased dramatically [2]. The World Health Organization now estimates that the 2013-2015 outbreak resulted in over 28,000 casualties and over 11,000 deaths in Liberia, Sierra Leone, and Guinea [3]. Fortunately, an international response has helped control the epidemic and early results from vaccination trials are encouraging [4]. But, even with the advent of anti-viral therapeutic drugs and vaccines as well as improving public health systems, the impossibility of eliminating the natural reservoirs of Ebola means that virus outbreaks will continue indefinitely. Another concern is the persistence of Ebola virus in the human body. Recent findings show that the virus can persist in ocular fluid in Ebola patients during their recovery [5] and in another study, Ebola RNA was detected in the semen of Ebola survivors nine months after the original onset of disease [6]. A case of sexual transmission of Ebola from a male survivor to his female partner has now been documented [7]. The recent Ebola epidemic underscores the need for supporting vaccination programs, better public health systems, and better disease surveillance. If pre-symptomatic diagnosis of an Ebola infection were possible, it could help manage future outbreaks and potentially save lives in developing as well as developed countries. A rapid diagnosis based on early altered host responses may be possible days before a test that is based on the emergence of viral RNA or protein and thus could provide a medically useful early warning when the risk of a positive test is extreme, such as to the family member of an infected patient or to a health care worker.
Existing evidence shows that Ebola infection results in dramatically altered host gene expression profiles in blood cells such as monocytes or macrophages [8-11]. Many of these changes occur before the emergence of clinical symptoms and well before the accumulation of high titers of virus in the blood. Several labs have published pioneering studies of Ebolainfected macaques [8,10] or cultured human cells [9,11]. We first surveyed publicly available ZEBOV infection studies and integrated this RNA data into a complete expression time course. Focusing on the first 24 hr postinfection we asked if host changes resulting from ZEBOV infection could be distinguished from those derived from three other pathogens: malaria, rhinovirus, and influenza. Next, we identified those early genes that were predicted to encode secreted or membrane bound proteins since these would be more readily assayed in peripheral blood. We then identified pairs of up- and down-regulated genes with high classification power for detecting infection or for distinguishing Ebola infection from infections by other pathogens (malaria, rhinovirus, influenza) that initially present with similar symptoms. As few as single pair of markers that have inverse expression profiles in two samples (disease versus normal; pathogen 1 versus pathogen 2) can be found that classify the samples into these two states [12-15]. Finally, since early reports have outlined selected changes to mammalian physiology after infection by ZEBOV [11,16-20] we used our integrated time course data to illustrate in detail the broad pattern of immune activation which occurs early but which is followed by profound immune suppression later. We suggest that the early candidate marker RNAs or proteins we described, if confirmed with further work, could become the basis for a pre-symptomatic blood diagnostic test. Because no data is publicly available from blood samples from all infection stages from a human infected with ZEBOV, we recognize the limitations of our approach but present it here as a step towards the goal of early diagnosis.
Materials and Methods
Sources of data and creation of a complete blood gene expression profile after ZEBOV infection
In order to establish a complete time course of the changing pattern of gene expression in blood after ZEBOV infection we re-analyzed and integrated differentially expressed genes (DEGs) from four studies: from macaques infected in vivo or primary human macrophages that were infected in vitro as shown in figure 1. To the extent possible we applied uniform data reduction methods as recommended for the respective microarray platforms. The macaque studies provided microarray gene expression data from 24 hours through 144 hours post-ZEBOV infection and were derived from the Gene Expression Omnibus (GEO) accessions GSE8317 [8] and GSE24943 [10], while the human studies provided data from 1 hour through 24 hour post-ZEBOV infection and were derived from GSE31747 [11] and GSE24125 [21]. The scope of these source studies varied, but we only re-examined data derived from human macrophages infected in vitro or peripheral blood mononuclear cells, infected in vivo, not other cell types or data from drug-treated cells. The four ZEBOVinfection studies we reviewed as well as studies of influenza, rhinovirus, and malaria infections are diagrammed in Supplementary figure 1 and listed in table 1. All of the re-analyzed data from the current study was deposited in the GEO database under accession GSE83331.
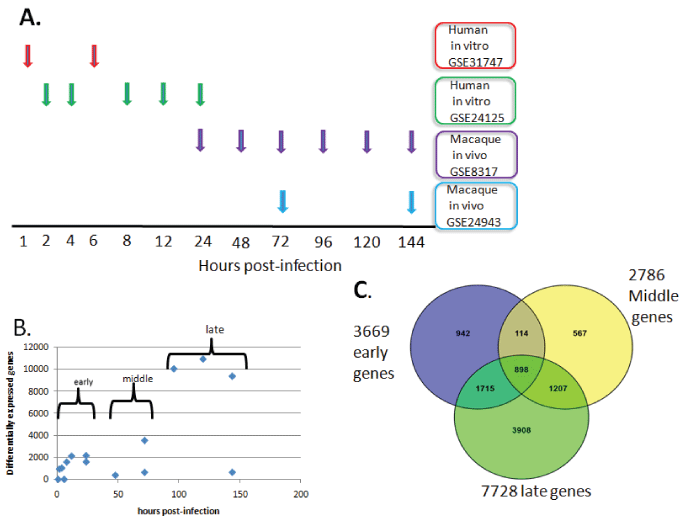
Figure 1: Outline of the human and macaque ZEBOV infection studies utilized for this analysis. Microarray data was derived from four published studies that encompassed the entire infection cycle.
A. Red, green, purple, or blue arrows indicate the microarray profiling times that were selected from two studies of human macrophages infected with Ebola virus-Zaire (ZEBOV) or two studies of macaques infected with ZEBOV; B. The number of differentially expressed genes (p ≤ 0.05, fold-change ≥ ± 1.5) from the studies in A., denoted with blue diamonds, were plotted as a function of time post-infection with ZEBOV. Early (0-24 hours post-infection), middle (48-96 hours post-infection), and late (96-144 hours post-infection) differentially expressed gene groups are shown; C. Overlap between early, middle, and late differentially expressed genes. Of the 3669 early genes, a substantial number overlapped with the middle (1012) or late (2613) study periods.
For GSE8317 [8], peripheral blood mononuclear cells from ZEBOV infected macaques were profiled by microarray hybridization before infection and at 1, 2, 3, 4, 5 and 6 days post-infection. Between 2 and 11 microarray samples from different animals were available at each day. A total of 15 pre-infection and 33 post-infection unique profiles were reanalyzed. We derived the DEGs for groups of animals at each time point before (pre-bleed) and after infection. GSE8317 contained a microarray having 37,632 probes representing approximately 18,000 human genes measuring macaque gene expression via the significant sequence homology between them. The ‘VALUE’ column in .txt files containing the log2 ratio of Cy5 (treatment channel)/ Cy3 (control channel) was imported into Agilent’s GeneSpring GX software for analysis after median baseline transformation.
In GSE24943 we re-examined gene expression data derived from peripheral blood mononuclear cells from macaques that were infected with ZEBOV and followed for longer times by Yen et al. [10]. The DEGs were derived from 4 pre-infection and 10 post-infection profiles from 4 animals that were not part of the investigational drugs that were tested as part of this study. DEGs were calculated by comparing post-infection with pre-infection microarray gene expression data for each animal. Three microarray profiles were available for 3 days post-infection, four were available for 6 days post-infection, and one data set was available from 7, 8, or 9 days post-infection. GSE24943 and GSE24125 contained twochannel data where the Cy3 (green dye channel) was the universal human reference and Cy5 (red dye channel) was the Ebola infected or mock treated samples. The lowess smoothing normalization was applied to background corrected median signal intensity values from both channels of these datasets so as to exclude spurious dye bias associated variations in the data before setting the median baseline.
Data from primary human macrophages that were infected with ZEBOV was available from two sources: GSE24125 [21] and GSE31747 [11]. In the former study, primary macrophages were isolated from 2 donors and microarrays were obtained with 7 control profiles and 7 infected profiles at 2, 4, 8, 12, and 24 hours, post-infection. From the latter study, microarray data was derived from 6 control profiles and 6 infected profiles at 1 hour and 6 hours post-infection. DEGs were derived at each time-point as for the non-human primate studies. GSE31747 contained Affymetrix microarrays (platform: HGU95Av2) and were summarized to the probe-level using robust multi-array normalization before setting the median base line using GeneSpring GX software. Due to the small differences in the means of Ebola infected vs. mock stimulated samples in GSE31747, the moderated t-test was applied to data as previously described in [22] to derive DEGs using asymptotically computed p-values at a cut-off of p ≤ 0.05 with a fold-change ≥ ± 1.5 without any multiple hypothesis testing correction. The unpaired student’s t-test was used for deriving DEGs from the remaining datasets at a cut-off of p ≤ 0.05 and fold-change ≥ ± 1.5.For deriving DEGs, Benjamini-Hochberg multiple testing correction was not applied in any of the four original studies, nor did we apply it in the current study at any time point since it excluded all or nearly all DEGs. We then combined these genes into groups that were expressed at early (0-24 hr), middle (48- 72 hr) and late (96-144 hr) times post-infection as shown in Supplementary figure 2.
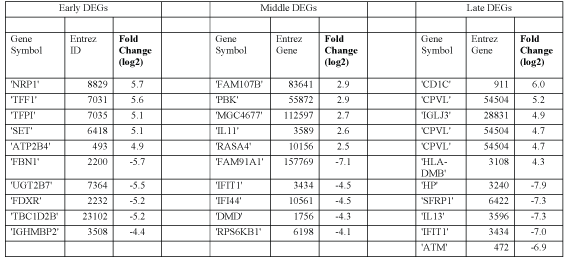
Figure 2: The most highly differentially expressed early, middle, and late stage ZEBOV genes
Malaria infected blood profiles were derived from dataset GSE5418, excluding the chloroquine data [23]. The original data was summarized, quantile normalized, and a median baseline was set at the level of individual probes. Differentially expressed genes were then derived using permutatively computed p-values at a cut-off of p ≤ 0.05 and fold-change ≥ ± 1.5 without any multiple hypothesis testing correction. The influenza (H1N1, strain A/WS/33, ATCC IV-1520) and rhinovirus (RV16) profiles were derived from dataset GSE71766 [24] and the differentially expressed genes (listed in supplementary file 5 of the original publication) were used for comparison with differentially expressed genes from Ebola and Malaria.
Deriving a master gene expression time course
We operationally defined the early phase of ZEBOV infection from 1-24 hours, the middle phase as 48-72 hours, while the late phase was 96-144 hours. The early phase combined data from GSE31747, GSE24125, and GSE8317 while the middle and late periods were based on data from GSE8317 and GSE24943. After removing duplicate entries, the ZEBOV early genes were compared to data from malaria infected blood [23] or genes that were differentially expressed after human bronchial epithelial cells were infected with rhinovirus or influenza virus [24].
Identifying secreted and membrane-associated proteins
In addition to blood derived mRNAs, abundant secreted or membrane associated proteins could be practical targets for early detection. Thus, mRNAs that are predicted to encode secreted proteins or proteins that are associated with the cell membrane were identified from the list of early differentially expressed mRNAs by searching the 3669 early mRNAs against three databases including information for cellular location of gene products: the Gene Ontology Cellular Consortium [25], The Human Protein Atlas [26]; and a database of subcellular protein localization ‘Compartments’ [27]. We identified mRNAs that encoded secreted proteins, by using the keywords “Extracellular or Vesicle”, “Secreted Protein”, and “Extracellular” for the Gene Ontology, Human Protein Atlas, and the Compartments searches. For membrane proteins, “plasma membrane” was also used for all three databases. The search results were curated manually for false positives and negatives, yielding a total of 581 mRNAs likely to encode secreted or membrane-associated proteins (Supplementary figure 3).
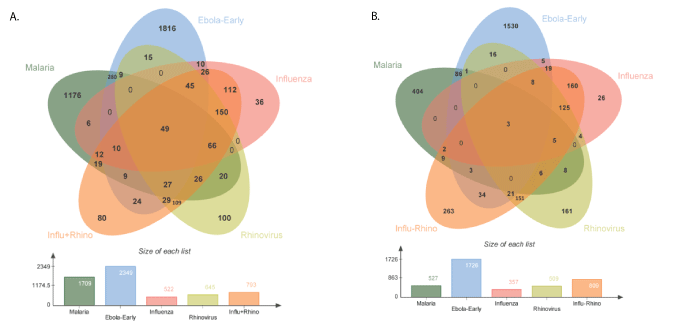
Figure 3: Comparison of ZEBOV early genes to differentially expressed malaria, rhinovirus, or influenza genes.
A. Up-regulated ZEBOV early genes compared to malaria, rhinovirus, and influenza.
B. Down-regulated ZEBOV early genes compared to malaria, rhinovirus, and influenza.
Calculation of top scoring marker pairs
Microarray data from 5 GEO datasets (Malaria, GSE5418; Ebola, GSE8317, GSE31747, GSE24125; Influenza, Rhinovirus & both viruses, GSE71766) were merged to identify 6795 genes in common. The normalized probe signal intensities were averaged per gene prior to deriving top scoring marker pairs. Top scoring pairs (TSP) analysis was performed using the R package provided by Bioconductor (ver. 3.2) ‘tspair’ [28] and k-Top scoring pairs (k-TSPs) were derived using the ‘ktspair’ R package [29]. The k-TSP analysis was based on expression data derived from early time points from both human macrophages and macaque PBMC after ZEBOV infection (0, 1, 2, 4, 6, 8, 12 and 24 hours), human bronchial epithelial cells (BEAS-2B) after 2, 4, 8, 12 and 24 hours post infection with influenza, rhinovirus and both viruses (group: RVIV) and human PBMC from individuals exposed to malaria. The TSPs score was calculated with ‘tspcalc’ function and evaluated by a permutation test using function ‘tspsig’. In this test, the p-value was computed based on a null TSP score distribution calculated by permuting the group labels 1000 times. Supplementary figure 4 summarizes the results obtained from ‘k’ top scoring pairs derived using the ‘ktspair’ R package. The accuracy is estimated by using the listed kTSPs per comparison and computing the performance of the group using a 15 fold cross-validation (functions: ‘cv2’, ‘ktspcalc2’ within the ‘ktspair’ R package).
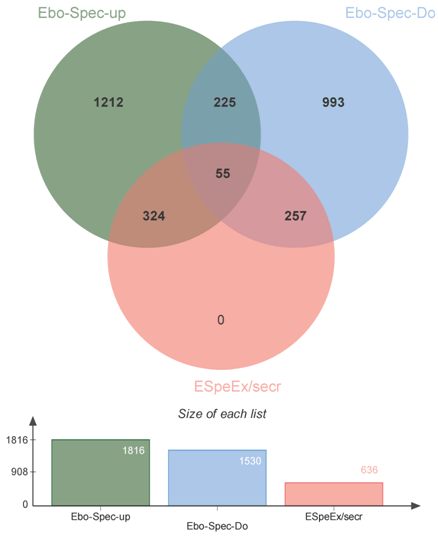
Figure 4: Identification of ZEBOV specific up and down regulated probes that encode membrane associated or secreted proteins derived from microarray probe data. 324 probes uniquely identify host genes encoding membrane or secreted proteins that are up regulated while 275 probes identify down regulated genes for membrane or secreted proteins. ‘Ebo-spec-up’: Ebola specific up regulated genes; ‘Ebo-specDo’: Ebola specific down regulated genes; ‘ESpeEx/secr’: Ebola specific genes that encode extracellular, membrane-associated or secreted proteins. Some microarray probes (225, 55) were scored as both up and down regulated are derived from cross hybridization and were not filtered out before this analysis.
Pathway analysis
Up- or down-regulated DEG from early, middle, and late stages of ZEBOV infection were mapped against biological processes compiled by the KEGG consortium [30,31], BioCarta [32], PANTHER [33,34] and REACTOME [35,36] pathways using the pathway enrichment tool embedded within the DAVID suite of programs (Database for annotation, visualization and integrated discovery; https://david.ncifcrf.gov/home. jsp) [37-39]. To identify those DEGs that were dysregulated after ZEBOV infection for a given biological pathway, a modified Fisher’s exact test was used to compute a p-value of 0.1 using DAVID’s functional annotation toolbox. The DEGs for T cell and B cell signaling as well as Toll-like and NOD-like receptor signaling were then mapped onto KEGG pathways using Adobe Illustrator.
Results and Discussion
A blood gene expression profile from the beginning to the end of an Ebola infection
We identified host genes that are differentially expressed in infected blood cells during the course of an Ebola virus infection from four published studies as indicated in figure 1. We derived DEGs from two studies in which human blood-derived primary macrophages were infected in vitro with ZEBOV [11,21] and two studies in which macaques were infected with ZEBOV in vivo and peripheral blood mononuclear cells were profiled [8,10]. In the latter two studies, the infected macaques were followed until they became moribund and were euthanized. All data (including control profiles) was obtained from the Gene Expression Omnibus. We analyzed data from fifteen time points over the course of 144 hours as shown in figure 1A. As indicated in figure 1B, DEGs from eight time points from the start of the infections to 24 hours post-infection derived from human and non-human primate studies were combined into an early set while DEGs from 3 time-points from 48-72 hours were combined into a middle set, and DEGs from time points 96-144 hours were combined into a late expressed gene set. DEGs were re-derived from the primary data using final criteria of p<0.05 and fold-expression changes of log2 ± 1.5 as described in methods. Since the infection time-course data we modeled was based on known, relatively high multiplicities of infection and took place with controlled in vitro or in vivo conditions, it probably represents a faster overall infection time course than would be expected in natural infections. We obtained over 3669 genes that were differentially regulated up or down and expressed early, 2786 middle period genes, and over 7728 genes that were differentially expressed at late times. The substantial overlap between the early genes which were derived from human and macaque data and the middle and late gene groups (entirely derived from macaque data) is shown in figure 1C which validates the use of macaques to model human responses to ZEBOV and our use of this data. The thirty genes most differentially expressed (up or down) at these stages of the ZEBOV infection are shown in figure 2. The complete lists of up and down differentially expressed genes from early, middle, and late times post-infection are presented in Supplementary figure 2.
Comparison of Ebola with malaria, rhinovirus, or influenza profiles
Malaria and cholera are endemic in much of West Africa, and patients can present with early symptoms from these pathogens that are similar to Ebola including nausea, headache, muscular and joint pain. Thus, a simple point-of care test that discriminates between ZEBOV and other common pathogens would be quite valuable in rural clinics. Towards the goal of exploring the feasibility of a more broadly based differential diagnostic, we compared DEG from Ebola to DEGs that are specific for malaria, rhinovirus and influenza virus. (We were unable to find a suitable dataset for cholera.) A malaria profile was derived from infected blood [23] while profiles for rhinovirus 16 or influenza (H1N1 strain A) were obtained after infection of the human bronchial epithelial cell line BEAS- 2B [24]. Indeed subsets of ZEBOV early probes were found that do not overlap with the malaria, rhinovirus, or influenza virus profiles as shown in figure 3. Figure 3A shows the overlap of up-regulated ZEBOV genes while figure 3B shows the overlap with down-regulated ZEBOV genes. The specific up and down regulated genes identified in figures 3A and 3B are presented in detail in Supplementary figures 5 and 6, respectively. The implication that the host responses to a ZEBOV infection at an early time can potentially be distinguished from malaria and two common viruses suggests that a specific and selective diagnostic method may be feasible. The clinical presentation from malaria can vary with many factors including the age and medical or infection history of the subject, their general health, the stage of infection and other factors currently beyond the scope of our comparison. While many other endemic pathogens such as cholera or other viruses should be compared to the Ebola early genes, the current comparison represents a first step to a more comprehensive differential diagnosis assay. But the finding that specific blood cell mRNAs distinguish these common infections and the identification of pathogenspecific genes that encode blood proteins may stimulate future studies.
We next identified the subset of the ZEBOV early up- or downregulated genes that are predicted to encode secreted or membraneassociated proteins since these might be readily detected in an assay based on a simple blood sample. These proteins were predicted from the early, middle and late Ebola gene lists as depicted in figure 1C and compared to the Ebola-selective probes identified in figures 3A and 3B. Figure 4 shows 324 up-regulated and 257 down-regulated Ebola-specific early genes that encode membrane or secreted proteins. These predicted proteins are listed in Supplementary figure 3. By this filtering and comparison process, the predicted candidate Ebola-specific proteins include many familiar chemokines (CCL16, CCl7), extracellular matrix proteins (COL6A2, COL4A4), extracellular metalloproteases (MMP1, MMP2, ADAM17) and many interleukins (IL8, IL9, IL10, IL16, IL18). Combinations of these secreted host-derived proteins could be tested as appropriate targets for discriminating ZEBOV infected from uninfected blood samples.
Top scoring marker pairs for classification of early Ebola infection
Since pairs of markers having reciprocal expression between two different conditions can have great power to classify between two states (i.e., infected vs uninfected; infected with pathogen 1 vs pathogen 2) we derived top scoring pairs of markers that could simplify the classification of ZEBOV-infected blood from uninfected or an endemic infection such as malaria. Samples were classified using top scoring pairs of genes identified using the ‘tspair’ R package depending on their relative rank within the given dataset, as described in methods. Figure 5 A summarizes the top scoring marker pairs from Ebola-infected blood compared to uninfected blood or compared to infections from other pathogens. Figure 5B through 5G identified the marker pairs that best classify Ebola infection (group 1) from uninfected (group 2) or Ebola (group 1) compared to the other pathogens (group 2). Each panel shows the expression of one member of a gene pair for e.g. GRIP1plotted as a function of the expression of the other member of the pair POU2F1 with the dots colored red or blue depending on their group. The black line represents the classification boundary that distinguishes the two groups apart for a given marker pair. In figure 5B we derived top scoring pairs of markers that classify ZEBOV infected from uninfected blood and provisionally, ZEBOV from malaria (Figure 5C), ZEBOV from influenza infected cells (Figure 5D and 5E), ZEBOV from rhinovirus infected cells (Figure 5F), and ZEBOV from cells infected with both influenza and rhinovirus (group RVIV in figure 5G). If marker pairs such as the ones we propose in Fig. 5 can be validated with clinical samples, the cost and complexity of a diagnostic test could become simpler. The complete list of k-top scoring pairs, including those marker pairs that are predicted to encode secreted or membrane associated proteins are presented in Supplementary figure 4. These marker pairs were derived individually for every comparison, and with the exception of one gene pair (KDM6A-MT1X), they do not overlap significantly, and could thus be combined for more classification power in a blood based assay. The strong performance of these marker pairs in correctly discriminating infected from uninfected samples encourages us to repeat this calculation as better data from Ebola infections and blood gene or protein expression profiles from other pathogens that are endemic to West and Central Africa such as Cholera becomes available. We presented them here, since they offer specific hypotheses for follow-up and also because they may help the designer of a point-of-care device who needs the greatest sensitivity and specificity possible from a small number of diagnostic markers. Although we report top scoring pairs of markers based on expression data, we recognize that the corresponding proteins would need to be validated as the basis of a field-deployable assay.
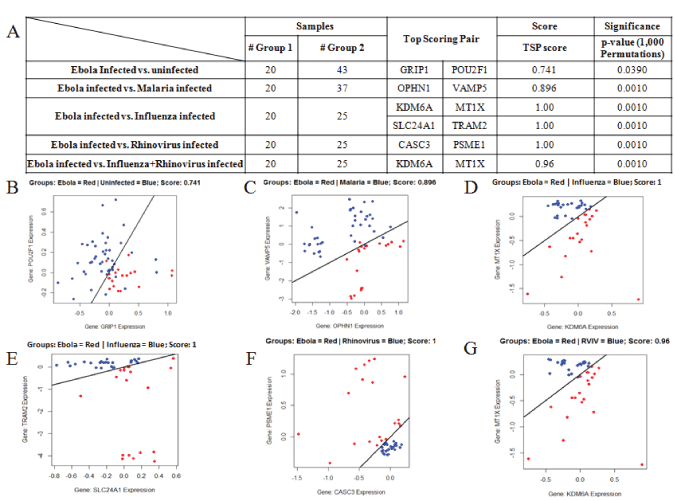
Figure 5: Top scoring pairs of markers that classify ZEBOV infections. Gene pairs with reciprocal expression profiles were identified that have the power to classify ZEBOV infected from uninfected samples or Ebola from other pathogens were derived and ranked as described in Methods. ;
A. Summary table shows gene pairs with high classification power for each of five diagnostic comparisons. The top scoring marker pair that classifies Ebola infected blood (group 1) from uninfected blood (group 2) is GRIP1 and POU2F1 B. Plot of expression data forGRIP1 and POU2F1 (blue : uninfected / red : ZEBOV infected); C. the best marker pair for identifying malaria infected blood from ZEBOV infected blood is OPHN1 and VAMP5 (blue: Malaria infected / red : ZEBOV infected) D. and E. the best marker pairs that discriminate ZEBOV-infected from influenza infected samples are KDM6A and MT1X (plotted) or SLC24A1 and-TRAM2 (blue: influenza infected / red: ZEBOV infected); F. the best marker pair that classifies Rhinovirus infected from ZEBOV infected cells is CASC3-PSME1 (blue: rhinovirus infected / red : ZEBOV infected); G. Marker pairKDM6A-MT1X also classifies cells infected with both rhinovirus and influenza virus (RVIV) from ZEBOV infected cells (blue: rhinovirus+influenzainfected / red: ZEBOV infected).
ZEBOV effects on immune response pathways
We mapped the differentially regulated genes we found fromthe early, middle, and late time groups to biological processes represented by KEGG pathways as shown in figure 6 where the colors represent p-values based on the number of genes that map to the pathway. Several immune system pathways stand out with a pattern of broad immune activation early in infection, followed by generally declining gene expression later. Supplementary figure 7 presents gene lists for the 32 pathways of Figure 6, suitable for detailed exploration of these pathways. To illustrate this in a gene-by-gene fashion we mapped the early up regulated genes along with the late down-regulated genes for two adaptive immune pathways, T cell receptor signaling and B cell receptor signaling and two innate immune pathways: Toll-like receptor signaling and nucleotide-binding oligomerization (NOD)-like receptor signaling in figure 7. Gene nodes that are early-up and late-down are all too common in essentially all of the principal adaptive and innate signaling pathways shown in figure 7. The data in Supplementary File 7 would support similar exploration of other pathways in the future. Supplementary figures 8A through 8D present these pathways in more detail.
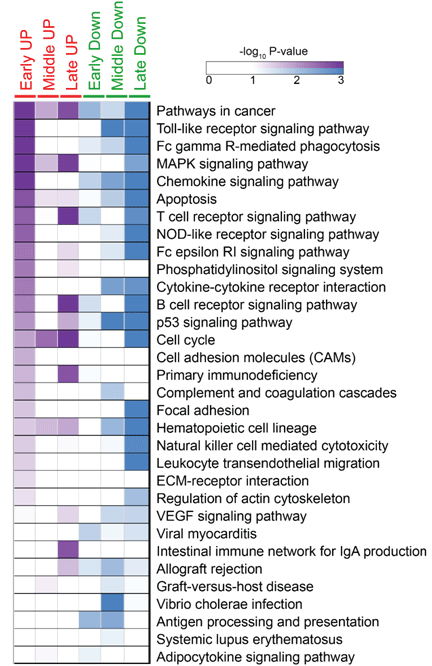
Figure 6: Pathways enriched due to Ebola infection. Pathways found to be enriched through a modified Fisher’s exact test implemented in DAVID from DEGs derived during the early, middle and late phases of Ebola infection. The enrichment is characterized depending on whether a DEG was either up (gradient: white to purple) or down (gradient: white to blue) regulated. Coloring for a given pathway being enriched within a given gene list (early, middle,late – up/down) is based on –log10 of the p-values obtained through Fisher’s exact test.
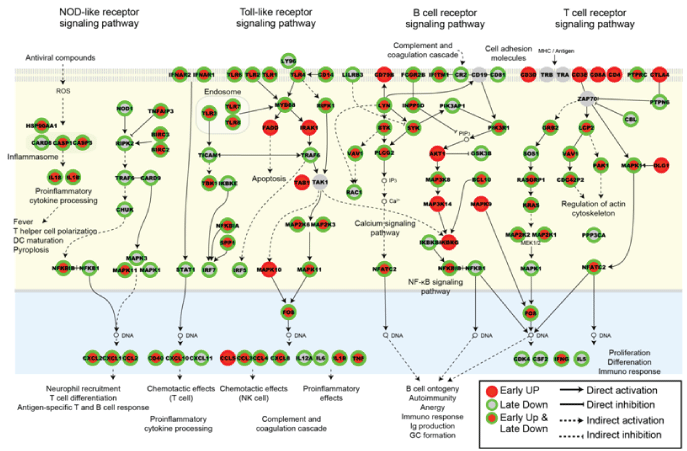
Figure 7: Adaptive and innate immune response pathways: active early but repressed late. Innate immune pathways for NOD-like receptor signaling and Toll-like receptor signaling and adaptive immune pathways for B cell or T cell signaling are plotted with colored symbols depicting early up regulation, late down regulation, or nodes that showed both early up followed by late down regulation. While a prompt and balanced immune response commences early (0-24 hr), late after infection (96-144 hr) this pattern is reversed at the mRNA level of a surprising number of key regulators.
Comparison with other studies
Our study included the data of Wahl-Jensen et al. [11]; who described gene expression in human macrophages at early times after infection in vitro with ZEBOV. These investigators identified many pathways or biological processes that were altered at 1 hour (43 pathways) or at 6 hours (60 pathways) post-infection including immune response pathways.Our detailed exploration of immune response pathways in figure 7 let us specifically confirm up-regulation of IL-10, TNFα, IL-8, CXCL1 and IL- 1β as previously noted in [11]. We also confirmed the strong suppression of innate immune responses which had previously described by Hartman et al. [18] after infection of liver cells with either ZEBOV or a variant harboring a mutation in the IRF-3 interacting domain of VP35. Hartman et al. noted inactivation of IRF-3 by the wild-type but not the mutant virus. We noted first up-regulation but then strong down regulation of IRF- 5, but as well the entire Toll-receptor signaling pathway resulting in the impaired production of IFN-α and IFN-β (Figure 7 and Supplementary File 8C). Figure 6 showed differential regulation of pathways for the cell cycle, p53 signaling and chemokine signaling confirming reports by Panchal et al. [20] derived from their studies of mouse models of ZEBOV infection. Figure 6 also confirmed the activation of T cell signaling and Toll-like receptor signaling as previously described by Barrenas et al. [16] in their study of an investigational vaccine trial in macaques infected with ZEBOV. Yen et al. [10] reported that mRNAs for CCL8/MCP-2increased and coagulation-associated genes TFPI and PDPN mRNA were decreased in macaques that were treated prophylactically with coagulation inhibitors and went on to survive ZEBOV infections. They suggested that these gene products might be useful as markers of survival post-ZEBOV infection. We have no evidence for differential up-regulation of CCL8 in our early data perhaps because the data from Yen et al was not part of our early gene group; it only contributed to our middle and late gene groups. Wauquier et al. [40] measured the levels of many cytokines and growth factors from individuals with fatal Ebola infections. Consistent with their results, we confirmed that the mRNAs for IL-1β, IL-8, MIP-1α, MIP-1β were up-regulated early, but in addition we noted that mRNAs for TNFα, CCL5(RANTES), CXCL1, MIP-2, CCL2, IL-18 and CD40 were also up-regulated early. More recently Ruibal et al. [41] described how the T cell inhibitory molecules CTLA-4 and PD-1 were much higher in patients who did not survive Ebola and lower in survivors. Consistent with this, our study that included data from macaques that did not survive ZEBOV infection showed a broadly based activation of the T cell response early, including elevated CTLA-4 mRNA as well as mRNAs for key components of the pathways that lead to normal T cell activation (PI3K-Akt; MAPK signaling; p38 signaling), as shown in Supplementary File 8A. But at late times, this pattern was reversed, consistent with T cell suppression.
General limitations and caveats
Our study was based on re-analysis of four separate data sets derived from human and macaques, infected with ZEBOV in vitro or in vivo, that were available in the literature by 2014. Even though the data came from infection studies of humans and macaques, many early DEGs were shared between species and many early DEGs continued to be expressed later during the infection cycle. We acknowledge that the velocity of gene expression changes in infected non-human primates or cultured cells may not be the same as in infected people. Thus we simply chose data early, from the first 24 hours of an infection time-course as a pragmatic approach to the problem. Other uncertainties contribute to when a person might first become aware that they are infected attributable to the virus (the dose, the portal of entry; the specific viral strain) or the particular individual (general state of health, the individual’s own innate and adaptive immune history and status, tolerance for discomfort, awareness of symptoms). We acknowledge that the gene pairs presented here represent a start towards a more advanced diagnostic method, and they might differ from the blood profile of host responses from a human patient during the entire infection cycle. For example, markers derived from mucosal surfaces might be under-represented here, since we studied data from fast-progressing controlled in vivo and in vitro sources. But if profiles of blood-derived mRNAs or proteins from the early stage of an ZEBOV infected human ever become available, our informatics workflow should be repeated and the list of host markers will improve. Limitations of the top scoring marker pairs include the fact these were based on microarray gene expression data, not (ideally) the levels of differentially expressed serum proteins. We acknowledge that the comparison of ZEBOV markers to those from other pathogens we presented was limited by the different cell types we compared (macrophages and peripheral blood mononuclear cells versus infected bronchial epithelial cells for influenza and rhinovirus) and thus important cell-type differences were missed. Again, deriving pathogen classification markers from blood protein profiles should be the next step in developing a more powerful early diagnostic test. A comprehensive diagnostic assay that might include host markers for other infections that are endemic in West Africa such as cholera, typhoid or other hemorrhagic fever viruses such as Lassa or Marburg would of course be valuable. The development and validation of such a test would entail considerable time, cost and complexity and it is beyond the scope of the current study. The influenza responses we compared to ZEBOV data were derived from bronchial epithelial cells not blood cells. We are aware that influenza blood profiles are now available [42] and these may be even more informative if this work is extended.
Value of pre-symptomatic markers in post-Ebola world
As long as man and ZEBOV co-exist, the need for an accurate, sensitive, and inexpensive diagnostic method will continue for the foreseeable future. Indeed, reliable negative results (i.e., not infected)will be as valuable as positive results for health care workers, family members of infected persons, and the general public in epidemic areas or anywhere in the world where an Ebola infection is suspected. About 20 outbreaks have occurred in West and Central Africa since 1976 [1] and more outbreaks will continue in the future. If a health care worker assigned to an EBOV outbreak receives a negative test result after work with infected or potentially infected patients, he or she would gain peace of mind and could return to work. If the test is positive, the worker could be isolated, commence supportive care, retest for EBOV or other relevant pathogens, and in the future hopefully start a course of anti-viral therapy. Indeed, in the West African epidemic, 800 health care workers were infected with Ebola and 500 of these died [43], emphasizing the need for early detection. A family member or anyone with even a casual contact with an infected individual who received a positive test could also take the above actions. Indeed, isolation alone might limit the spread of disease. For these reasons, once validated, a new diagnostic method that could identify an active infection as early as possible within the 21 day incubation period would be valued both in the developed as well as the developing world.
Conclusion
Even with the emergence of therapies and vaccines for Ebola virus, there will be an ongoing need for a specific, sensitive, and inexpensive point-of-care assay that can discriminate infected from uninfected individuals from blood samples early after a possible exposure. An assay based on host responses could be independent of other assays that detect the Ebola genome or gene products. Here we used a practical informatics workflow to identify markers from ZEBOV-infected blood derived cells that represent candidates for such an assay. The candidate markers should be improved, if data from a human infection time course study ever becomes available. We suggest that surprisingly few blood markers based on host responses may have enough power to identify not only infection by ZEBOV, but other common pathogens at the same time. Such an assay could always be followed up with conventional assays for viral protein or nucleic acid, as appropriate. While the recent Ebola epidemic has subsided, a new outbreak was recently reported from Guinea (Guilbert, K. March 18, 2016; Reuters) which underscores the need for a sensitive, accurate, and inexpensive point-of-care diagnostic method that could detect infections based on host protein responses early in the infection cycle.
Acknowledgement
This work was supported by the National Science Foundation (RAPD award 151330) as well as by support from the Institute for Systems Biology. We thank Kathie Walters and Jeff Boore for critical reading of the manuscript.