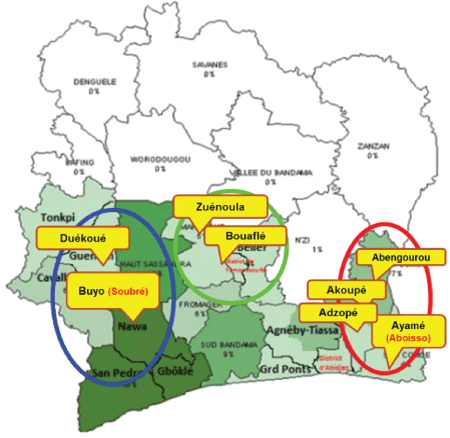
Figure 1: The three major cacao-growing regions in Cote d’Ivoire from which the plant samples analyzed in this study were collected. The western, central and eastern regions are indicated on the map as blue, green and red, respectively.
Nomatter Chingandu1 Koffie Kouakou2 Romain Aka2 Osman A Gutierrez3 Judith K Brown1*
1School of Plant Sciences, University of Arizona, Tucson, AZ 85721, USA*Corresponding author: Judith K. Brown, School of Plant Sciences, 1140 E. South Campus Drive, University of Arizona, Tucson, AZ 85721 USA, E-mail: jbrown@ag.arizona.edu
Cacao swollen shoot virus (CSSV) [Badnavirus, Caulimoviridae] causes swollen shoot disease of cacao (Theobroma cacao) L. in West Africa. Beginning in ~2002, tests once effective for detection of CSSV failed to detect virus in ~50-70% of symptomatic cacao plants in Ghana and Cote d’Ivoire, suggesting the possible emergence of an uncharacterized CSSV variant(s). Asymptomatic and symptomatic cacao and non-cacao plant samples from Cote d’Ivoire were subjected to polymerase chain reaction (PCR) amplification using eight different sequence-specific and/ or degenerate primer pairs with an expected size amplicon of 375-1100 base pairs. The frequency of PCR-amplification was variable, depending on sample-primer combination. Virus was not detectable in all symptomatic samples, despite characteristic CSSV-like symptoms, and several asymptomatic samples were CSSV-positive. Phylogenetic analysis using Maximum Likelihood of DNA sequences determined from the amplicons resulting from each primer pair resolved two to three groups, two that were closely related to previously reported CSSV isolates, and a third previously undescribed group. Based on the badnavirus species threshold at ≥ 80% pairwise nucleotide (nt) identity for the taxonomicallyinformative RT-RNase H region of the genome, analysis of a partial fragment corresponding to this locus resolved four CSSV groups, at 66-99% nt identity, among the PCR-amplifiable field isolates. Also, sequence analysis of as many as seven additional regions of the CSSV genome revealed extensive within-genome variability. These findings provide robust evidence for extensive genomic variation among multiple, divergent CSSV variants associated with swollen shoot disease symptoms in cacao in West Africa.
Badnavirus; Caulimoviridae; dsDNA virus; Emergent plant virus; Mealybug-transmitted virus; Pararetrovirus
Theobroma cacao (L.), or cacao (Malvaceae), is cultivated for the bean used to manufacture chocolate, confectionaries, and non-food products. Over 70% of the global supply of beans is produced in West Africa making it an essential crop for economic and food security [1]. The yield and quality of beans are reduced by feeding damage caused by insect pests, and by pathogens, including fungi [2] and plant viruses [3,4]. The Cacao swollen shoot virus (CSSV) [5] is the most economically-important among plant viruses known to infect the cacao tree. The virus was first reported in 1936 in cacao in Ghana [6]. It is endemic throughout West Africa, including in Cote d’Ivoire [7,8], Nigeria [9,10], Sierra Leone [11], and Togo [12,13], infecting many uncultivated species in the Bombacaceae, Malvaceae, Sterculiaceae, and Tiliaceae [14-16]. Further, outbreaks of CSSV in cacao have been correlated to the proximity of farms to native forest trees, and disease prevalence and rate of subsequent spread are influenced by elevation, precipitation, and temperature [17].
CSSV is classified in the genus Badnavirus (family, Caulimoviridae) [18,19]. The double-stranded, circular DNA genome of CSSV of approximately 7.0-7.3 kilo base pairs (kbp) in size, is encapsidated in a non-enveloped 128 × 28 nm bacilliform particle [20]. The badnavirus genome contains one discontinuity in the viral plus strand [21,22], the location of the priming site for reverse transcription of the DNA minus strand [19,23]. The CSSV genome encodes from four to six open reading frames (ORFs), which are named ORFs 1-4, and X and Y [8,21,24]. Although the function of many of the viral genes remain uncharacterized, ORF2 is known to encode a nucleic-acid binding protein [25], whereas, ORF3 encodes a polyprotein that is processed into movement (MP), coat protein (CP), and reverse transcriptase (RT), and an aspartate protease (AP) and ribonuclease H (RNase H) [24] that function in polypeptide cleavage and RNA hydrolysis, respectively.
Symptoms caused by CSSV infection of cacao trees range from mild to severe and vary seasonally, being most evident and severe on new flush growth that develops following each rainy season. Foliar symptoms develop predominantly on newly developing and young leaves, manifest as red and green vein-banding, or yellow- or white-green mosaics. New vegetative growth developing from the base of the tree or the trunk, referred to as shoots, develop swellings caused by viral-induced phloem proliferation [26]. Within 3-5 years after symptom development, infected trees undergo dieback, decline, and then death. Compared to uninfected trees, CSSV-infected trees produced fewer pods and beans, and both size and quality of beans are reduced [27].
The CSSV is transmitted in a semi-persistent manner by 14 mealybug species [28,29], but is not pollen or seed transmitted [30], despite evidence that it can be detected transiently in seed [31]. The long and short distance spread of CSSV is exacerbated by human-mediated exchange of virusinfected cacao cuttings.
Disease management has relied on replacement of infected trees with virus-free planting material, either grown from seed, or more recently, as seedlings [32-34]. Also, cross-protection using mild strain such as N1 or SS365B, showed initial promise for reducing the effects of CSSV severe strain 1A [35,36], however, a recent study found that spread of the severe CSSV strains had increased over time, resulting in deterioration of the beneficial effects of mild-isolate induced cross-protection, and leading to decreased life spans of trees [37]. Chemical control of mealybug vectors has shown minimal promise for reducing rates of virus spread [38,39]. However, the expansion of cacao farms to previously uncultivated locales, while at the same time abandoning diseased farms that can serve as a CSSV inoculum source has resulted in virus spread into new plantings by the mealybug vectors [1]. Long-term disease management has been attempted by breeding programs established during the 1940’s and ongoing to the present, to develop CSSV resistance. These efforts have exploited cacao germplasm introduced into West Africa from the Amazon Basin of South America, however, resistance even when effective for periods of time, has not proven durable [40-43]. Despite management efforts, virus infection has continued to be manifest as characteristic CSSV symptoms, or recently, by a rapid decline and death phenotype first observed in western Ghana during ~2000-2002, and then in eastern and western Cote d’Ivoire [44] and Togo [45] during 2003-onward. Whether the similar symptom phenotypes observed at different sites where outbreaks have occurred are associated with the same CSSV variant(s) has not been determined.
Previously, an enzyme-linked immune sorbent assay (ELISA) was widely used for CSSV detection in West Africa [10,46,47]. Recently, ELISA tests have failed to detect CSSV in approximately 25-30% of samples analyzed from symptomatic trees. To the present, there is no serological or molecular diagnostic test available for comprehensive CSSV detection.
Seven complete CSSV genome sequences have been deposited in the NCBI GenBank database. They range from 7006-7297 bp in size and collectively, share 71-98% nucleotide (nt) identity (authors, unpublished data). Based on alignment of the seven genome sequences, PCR primers have been designed that amplify a fragment of CSSV ORF1 [48] or ORF3 [49]. However, neither primer set was found capable of detecting CSSV in all symptomatic leaf samples tested from Ghana [30,50], Cote d’Ivoire, or Togo [13,49]. An alternative practice used for CSSV detection in exotic germplasm has been applied in quarantine units, and uses grafting of scions from suspect plants onto the CSSV-susceptible ‘Amelonado’ rootstock, followed by periodic inspection of indicator plants for threeyears, post-grafting for evidence of CSSV symptoms [51], overall, a timeconsuming, expensive, and impractical approach.
The increasing spread of CSSV, including both isolates associated with the classical CSSV symptom phenotypes, and the recently discovered ‘severe decline’ phenotypes has made the development of effective diagnostic tests essential, to enable the epidemiological studies that rely on understanding the identity and distribution of CSSV variants throughout the region. Indeed, sequencing of PCR amplicons representing a variable region found in ORF3 that encodes the viral movement protein, has resolved six and nine CSSV groups in Cote d’Ivoire and Ghana, respectively [49,50]. Although this apparently, highly divergent coding region is not considered taxonomically informative at the species level, results underscore the greater-than expected molecular variability observed among CSSV isolates, and suggests the possibility that a number of as yet undescribed species and/or strains may be associated with distinct CSSVlike symptoms occurring in cacao plantings throughout West Africa.
The objective of this study was to design degenerate or non-degenerate primer sets for CSSV detection that were feasibly based on sequence alignments of the seven available CSSV genome sequences. Molecular testing focused on cacao samples collected from trees in CSSV-like outbreak areas that exhibited a range of symptom phenotypes occurring in cacao plantations throughout eastern, central, and western Cote d’Ivoire. Two previously published primer pairs designed for molecular detection of CSSV [48,49], a pair designed for ‘universal’ badnavirus detection [52], and five primers designed herein around selected different genomic coding and non-coding regions, or ‘loci’, were evaluated for the ability to detect CSSV by polymerase chain reaction (PCR) amplification. Annotation of the amplicons indicated that some sequences were non-viral, and often of cacao host origin, indicating occurrences of viral-plant host sequence homology and therefore lack of primer specificity. Those annotated as CSSV-like and analyzed with their respective genomic loci with respect to pairwise distances and phylogenetic relationships, revealed extensive between-genome variability resulting in multiple molecular variants.
Materials and Methods
Samples of leaves and/or swollen shoots were collected from 91 symptomatic and asymptomatic cacao trees in the three major cacao growing regions of central, eastern, and western Cote d’Ivoire, experiencing recent and/or long-term CSSV outbreaks (Figure 1), and from experimentally inoculated, young, symptomatic cacao plants maintained at the Centre National de Recherche Agronomique (CNRA), Côte d’Ivoire, in 2012. An additional 33 samples were collected from suspect endemic CSSV hosts, representing 15 species, growing near cacao plantations. The plant samples were preserved in glycerol, and stored at 4°C. Total DNA was isolated from 100 mg of leaf or shoot tissue using the cetyl trimethylammonium bromide (CTAB) method [53]. The final pellet was dissolved in 100 µL of low TE buffer (10 mM Tris-HCL (pH 7.5), containing 0.1 mM EDTA (pH 8.0), and stored at -20°C.
Figure 1: The three major cacao-growing regions in Cote d’Ivoire from which the plant samples analyzed in this study were collected. The western, central and eastern regions are indicated on the map as blue, green and red, respectively.
Five primer pairs were designed based on a multiple sequence alignment of seven available full-length CSSV genome sequences: Accession numbers AJ534983, AJ608931, AJ609019, AJ609020, AJ781003, JN606110, and L14546. The sequences were aligned using MUSCLE [54] implemented in CLC Sequence viewer 7.5 (http://www.clcbio.com/ products/clc-sequence-viewer). Four primer pairs were designed within ORF3 and one pair on the non-coding intergenic region. The coordinates and approximate size of PCR amplicons are provided in (Table 1 and Figure 2). Three previously published primer sets included for comparison were those designed to target the movement protein (5’-end of ORF3) [49] or a fragment of ORF1 [48], and the degenerate ‘general’ badnavirus Badna FP/RPprimers [52] (table 1, figure 2), herein, referred to as ORF3A, ORF1, and Badna, respectively. To assess DNA quality, eight samples were selected for PCR amplification of a 2-kbpplant mitochondrial DNA intron located within the nad4 gene, between the exons 1 and 2 [55].
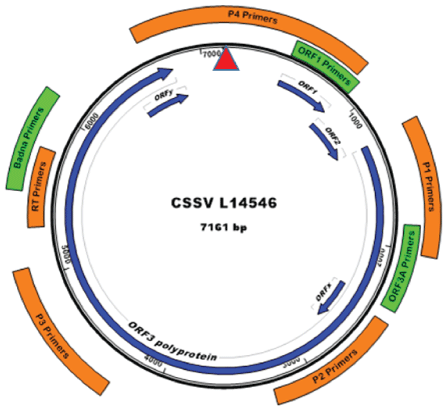
Figure 2: The genome sequence map of Cacao swollen shoot virus (CSSV) showing approximate primer coordinates, based on the Genbank CSSV isolate Accession number NC_001574. The previously published primers, Badna [52], ORF1 [48], and ORF3A [49] were designed to target regions denoted in solid green. The locations of primers designed herein are shown in solid orange. The red arrow indicates the position of nucleotide coordinate one, based on the convention established for badnaviral genome sequences.
| Primers forward (F), reverse (R) | Primer pairs 5’– 3’ orientation | Primer coordinates | Primer Ta°C | Expected size (bp) |
| RT_F | AACGACAACACTGAAAAGGA | 5325 - 5344 | 56 | 421 |
| RT_R | GTGCCCAAAAATTCAATCTC | 5727 - 5746 | 56 | |
| ORF3A_F | GTYRTACCRRAYAYYATGATGAC | 1848 - 1870 | 55 | 532 |
| ORF3A_R | GTTYCCRTTRSYRGAYTCYTCCCATAC | 2355 - 2380 | 55 | |
| ORF1_F | AACCTTGAGTACCTTGACCT | 498 - 517 | 56 | 375 |
| ORF1_R | TCATTGACCAACCCACTGGTCAAG | 849 - 872 | 56 | |
| P1_F | RGCAGCWGAARWGGCWAAGR | 1244 - 1263 | 56 | 774 |
| P1-R | TTDGGWGTRTTKGAYARYCK | 1999 - 2018 | 56 | |
| P2_F | ACDGGHTGGGRMRRYGATRA | 2461 - 2480 | 56 | 804 |
| P2_R | TRTCYTTKATRTTGTKGCADGT | 3244 - 3265 | 56 | |
| P3_F | ATNMHRGTCCARCAGCAGCC | 4089 - 4108 | 56 | 1042 |
| P3_R | TTDATGGGCTTRTCTTCWAT | 5112 - 5131 | 56 | |
| P4_F | TGGCAACDGAACATGCCATCTC | 6585 - 6606 | 56 | 1123 |
| P4_R | TGGTTGTTGGTCACTTTACT | 528 - 547 | 56 | |
| BadnaFP | ATGCCITTYGGIAARAAYGCICC | 5513 - 5536 | 50 | 577 |
| BadnaRP | CCAYTTRCAIACISCICCCCAICC | 6066 - 6090 | 50 |
Table 1: Primer pairs used for PCR-amplification of eight Cacao swollen shoot virus genomic regions. The primer coordinates are based on the CSSV reference sequence, Genbank Accession number NC_001574.1
The circular DNA present in purified field sample DNA was enriched using rolling circle amplification (RCA) and phi29 DNA polymerase [56] (Templiphi RCA kit, GE Healthcare Bio-Sciences, NJ, USA) according to manufacturer’s instructions with modifications as described [57,58].
The RCA product was used as template for PCR amplification. Reactions contained 10.5 µL nuclease-free water, 12.5 µL RedTAQ mix (Sigma-Aldrich, St. Louis, MO, USA), 0.2 µM of each primer, and 1 µL RCA template in 25 µL. Cycling parameters were: initial denaturation for 2 min at 94°C, followed by 35 cycles each of denaturation at 94°C for 30s, annealing at 50-56°C for 30s, extension at 72°C for 1 min/kbp, with a final extension at 72°C for 10 min. Parameters for Badna primers were as previously described [52]. The expected amplicon sizes were verified by agarose gel (0.8%) electrophoresis, in Tris-acetate EDTA buffer, pH 8.0. The bands were visualized by GelRed staining (10 µL/mL) (Biotium, Aurora, CO, USA).
Only PCR amplicons of the expected sizes were ligated into the pGEM T-Easy plasmid vector (Promega, Madison, WI, USA), followed by transformation into Escherichia coli DH5α bacterial cells, according to manufacturer’s instructions using blue-white selection. Colony PCR was carried out in1X PCR buffer, 0.5 U Platinum Taq polymerase (Invitrogen), 0.2 mM dNTP mix (Sigma-Aldrich, St. Louis, MO, USA), 0.2 µM each of M13 forward and reverse primers, and nuclease- free water, in a final volume of 50 µL. Amplicon size was verified by agarose gel electrophoresis as described above. Two to three plasmids per sample, each bearing the expected size insert, were subjected to bi-directional Sanger capillary DNA sequencing at the University of Arizona Genetics Core sequencing facility (Tucson, AZ).
The DNA sequences were assembled using SeqMan Pro Software Lasergene version 11 (DNASTAR, Madison, WI), and annotated using BLAST2GO software [59]. The CSSV sequences were aligned using MUSCLE [54] implemented in CLC Sequence viewer, version 7.5. To reduce the number of sequences used for the analyses, those sequences sharing 100% identity, i.e. ‘haplotypes’, were removed using FaBox v1.41 software [60]. Representative haplotype sequences for each CSSV genomic locus were deposited in the NCBI GenBank database, and assigned the Accession numbers KY473626 - KY473897.
The pairwise nt identity was calculated for each group of amplicons, per primer pair or genomic locus, using Sequence Demarcation Tool software (SDTv1.2) [61]. The amplicon sequences representing the partial RT-RNase H locus were grouped with a basis in ≥ 80% shared nt identity
Phylogenetic analysis was carried out using Maximum Likelihood (ML), implemented in MEGA6 [62] with 1000 bootstrap iterations, and the nt substitution model [62] predicted to have the lowest Bayesian Information Criterion score.
Analyses of the PCR amplicon sequences obtained using the combined results for each primer set, indicated CSSV was detectable in 56 cacao and 13 non-cacao plant samples (from a total of 69) of the 124 samples tested. However, the ability of each primer set to produce an amplicon of the expected size was highly variable, depending on the sample. Certain primers did not produce a CSSV-amplicon from samples otherwise confirmed to be CSSV-positive by PCR using other primer pairs, indicating that the PCR-amplification was generally inconsistent among the primer sets. The PCR-amplification of a CSSV-like sequence by one or more of the eight primer pairs (Table 2) indicated that 69 of 124 samples were positive for CSSV detection e.g. verified for each sequence by BLASTn analysis. The number of amplicons obtained from the 124 field samples using the eight primer pairs ranged from 24 to 52, representing an amplification rate ranging from 19 and 42%. Among the 69 samples that were confirmed CSSV-positive by one or more primer set, virus was found to be detectable in only35 to 75% samples.
| RT | ORF3A | ORF1 | P1 | P2 | P3 | P4 | Badna | |
| I | 46/69 (67%) | 41/69 (59%) | 24/69 (35%) | 29/69 42% |
34/69 49% |
41/69 59% |
52/69 75% |
35/69 51% |
| II | 46/124 (37%) | 41/124 (33%) | 24/124 (19%) | 29/124 (23%) | 34/124 (27%) | 41/124 (33%) | 52/124 (42%) | 35/124 (28%) |
| III | 2/46 | 2/41 | 1/24 | 0/29 | 1/34 | 1/41 | 6/52 | 3/35 |
Table 2: Frequency of polymerase chain reaction amplification of a fragment of the Cacao swollen shoot virus (CSSV) genome using the eight primer pairs. Rows/Panels I and II show the number and percent of samples amplified by each primer pair, in relation to the 69 samples confirmed positive for CSSV, and to the 124 total samples tested, respectively. Panel/ Row III indicates the number of non-cacao plant samples positive for CSSV, in relation to samples positive for CSSV using each primer pair
Among the primers tested, P4, which targeted the CSSV noncoding intergenic region yielding a 1,123 bp product, had the highest amplification frequency, at 42% (52/124), whereas, the PCR primers designed to amplify an expected size product of 421 of the CSSV conserved RT region (located in ORF3), showed the second highest frequency, at 37% (46/124).
The previously published ORF3A primers and the P3 primers (herein), amplified CSSV at a similar frequency of 33%, or 41 of 124 samples. The ORF3A primer target is a 532 fragment of the ORF3-movement protein gene, located at the nt coordinates 1848 – 2380 (GenBank Accession NC_001574.1), compared to P3 that was designed to amplify a different ORF3 fragment corresponding to the pepsin-like aspartate protease region. When the Badna primers were tested, only 28% (35/124) of field samples were CSSV positive. The Badna primers have been reported to be ‘universal’ or genus-specific, having been designed by taking into account all badnaviral genome sequences available in public databases at the time. The 577 bp Badna amplicon represents about half, or 46.9%, of the RTRNase H locus sequence. This 577 bp fragment can be amplified from divergent variants using these primers because they and the region they amplify represent the most conserved region within the RT-RNase H locus that it selfis ~1,230 bp in size. This 1,230 bp region of the CSSV and other badnaviral genomes has been ratified by the International Committee on Taxonomy of Viruses (ICTV) for species demarcation in the genus Badnavirus [19]. Although the fragment amplified here represents only about 47% of the RT-RNase H domains combined, it has been widely used for provisional badnaviral species classification, nonetheless [50,63-66]. In lieu of having the complete RT-RNase H sequence for isolates studied herein, the use of this region for pair wise nucleotide identity analysis to provisionally classify the isolates to species followed these latter examples.
Amplification of CSSV using the P2, P1, and ORF1 primers yielded approximately one-fourth or less of the field isolates tested, at 27% (34/124), 23% (29/124), and 19% (24/124), respectively. The P1 and P2 primers targeted the ORF3 5’-end, yielding amplicons of 774 bp and 804 bp in size, respectively. Also, the P1 primers amplify the region located between nt coordinates 1244 - 2018, which overlaps with the target region of the previously published ORF3A primers [49]. The P2 primers amplified a fragment flanked by nt coordinates 2,461 - 3,265, in a region that harbors no predicted functional protein domains in NCBI Conserved Domain Database searches [67]. The ORF1 primers directed amplification of a 375 bp region of ORF1 that contains a badnavirus-specific domain of unknown function, DUF1319 [67] present in all seven available genomes.
To determine whether the inability of the PCR primers to amplify CSSV from otherwise CSSV-symptomatic samples could be due to DNA quality, eight samples were selected for testing by PCR-amplification with nad4 primers [55], based on poor CSSV amplification. Four of the eight samples did not yield a product regardless of CSSV primer pair, and four had been amplified by only one or any two CSSV primer pairs. Results using the nad4 primers indicated that all eight samples yielded the expected 2 kbp amplicon (data not shown), confirming that the DNA could be used for successful PCR amplification of plant genes, and therefore should also have produced CSSV amplicons given sufficient virus titer.
The GenBank database BLASTn search for the 69 amplicon sequences obtained revealed robust match(es) with one or more CSSV sequences, at an e-value of 0 and ≥ 87% similarity scores. The only exceptions were sequences from five samples amplified with Badna, ORF3A, and P3 primers, which had similarity scores of 69-76%, e-values of 8e-61 to 1e- 102, and only 30-70% coverage.
One or more primer pairs successfully amplified CSSV-like or other badnaviral sequences froms even non-cacao plant species. The plant host species and successful primer(s) were for: Carica papaya (Badna,P4,RT), Ceiba pentandra (all except P1), Commelina erecta (Badna), Dioscorea cayenensis (P4), Spigelia anthelmia (P4), Tapinanthus bangwensis (P4), and Xanthosoma maffafa (ORF3A,P4). Among these endemic plant species, C. pentandra has been previously reported as a host of CSSV in mealybug transmission studies [15].
All eight PCR primers, except ORF1, produced at least some amplicons that when sequenced, were annotated as cacao genomic DNA. The BLASTn results indicated that most of the cacao sequences shared their highest similarity score(s) with T. cacao sequences. Examples of cacao sequence hits included the DNA/RNA polymerase super family, gag-prolike proteins, receptor kinases, retrotransposons, reverse transcriptase, transport proteins, or T. cacao uncharacterized proteins. Perhaps surprisingly, among the primers tested, the Badna primers amplified the greatest number of cacao sequences, at 48%. Also, the Badna primers amplified a 577 bp fragment that was annotated as Dioscorea bacilliform virus (DBV) or Banana streak Uganda E virus (BSUEV). The latter amplicons were associated with four cacao samples that also yielded CSSV amplicons with six of the eight primers, suggesting that they co-infected cacao with CSSV. Also, BLASTn searches identified DBV-like badnavirus sequences in X. maffafa (n = 2) and D. cayenensis (n = 1), and a BSUEVlike sequence in T. bangwensis (n = 1), with similarity search scores at 73–82% for DBV and 73% for BSUEV, with 48-74% coverage and e-values of 1e-53 to 6e-51.
Analysis of the RT-RNase H partial fragment delimited by the Badna primers using ≥ 80% nt identity species threshold indicated a shared nt identity, at 68-99%, among all isolates for which the fragment could be amplified. However, in the absence of a complete ~1230 bp RT-RNase H sequence, the analysis was not considered taxonomically informative, albeit, eventually it will become possible to determine if the result is taxonomically consistent with the complete locus predictions.
Pairwise distance analysis of CSSV sequences amplified with the eight primer pairs indicated 66 – 99% shared nt identities. The P1, P4, and ORF1 primers yielded 63, 98 and 53 CSSV-like amplicons from the field isolates, respectively (Table 3). The loci delimited by P1, P4, and ORF1 primers were found to be equally divergent from each other by pairwise distance analysis, at 74 – 99% shared nt identity, however, the amplicons are not necessarily from closely related isolates, given the different amplification frequencies for primers P1, P4, and ORF1, at 23, 42 and 19%, respectively (Table 2).
| Primer / region | Amplicon sequences | Pairwise nucleotide identity |
| ORF3A | 85 | 68-99% |
| RT | 77 | 73-99% |
| P1 | 63 | 74-99% |
| P2 | 68 | 71-99% |
| P3 | 82 | 66-99% |
| P4 | 98 | 74-99% |
| ORF1 | 53 | 74-99% |
| Badna | 87 | 68-99% |
Table 3: Pairwise nucleotide identities of amplicon sequences determined from a fragment of the Cacao swollen shoot virus (CSSV) genome amplified using the eight primer pairs
The Badna and ORF3A primers produced 87 and 85 viral amplicons, respectively, which shared similar nt identity, at 68–99%. Pairwise nucleotide analysis for the Badna amplicons resolved four CSSV groups, at ≥ 80% species cutoff. Although the Badna primers amplify the partial region of the RT-RNase H used for taxonomy of badna viruses, the analysis delimiting four (SDT) groups represents four CSSV variants that are will most likely be characterized as different species. However, it will be necessary to consider the entire ‘informative sequence’ for each to test this hypothesis. Among these, Group I contained GenBank CSSV Accessions AJ609019, AJ608931, AJ609020, L14546, and AJ534983, whereas, Groups II and III harbored Accessions AJ781003 and JN606110, respectively, and Group IV contained previously unreported CSSV-like variants that were highly divergent from Groups I, II, and III isolates, at 73-77%,70-75%, and 71-75 %, respectively.
Based on pairwise identities, the P2- and RT-amplicons shared 71–99% and 73-99% nt identity, represented by 68 and 77 sequences, respectively. The greatest divergence occurred among the 82 P3-amplicon sequences, at 66-99% nt shared identity
Maximum likelihood analysis (>70% bootstrap; 1000 iterations) of the eight groups of amplicons resolved at least two clades per group, which also contained one or more CSSV GenBank reference sequences, and each clade was statistically supported (Figure 3). The CSSV-like sequences determined from the seven non-cacao samples grouped with sequences from cacao samples that clustered with available CSSV GenBank sequences representing cacao isolates from Togo, Ghana or Cote d’Ivoire.

Figure 3: Phylogenetic trees (ML)(1000 bootstrap iterations, >70% bootstrap) of Cacao swollen shoot virus (CSSV) sequences amplified using the eight primers, ORF1 (a), RT (b), Badna (c), ORF3A (d), P1 (e), P2 (f), P3 (g), and P4 (h). The CSSV sequences available in the NCBI- GenBank database are indicated by Accession number, in bold. Field isolates collected from the central, eastern and western regions are labeled using green, red and blue letters, respectively. Statistically supported clades are designated I, II or III. Sequences indicated by an asterisk (*) were determined from endemic, wild plant species. The partial genome fragment of Citrus yellow mosaic virus (Genbank Accession number AF347695) corresponding to the ORF3 priming region, was included as the outgroup (d). The use of the designations, Group A – F (d) was adopted from Kouakou et al. [49] and were used to compare to the current grouping system. The letter codes CI (Cote d’Ivoire), GH (Ghana), or TG (Togo) indicate the country from which cacao samples were collected
Phylogenetic trees reconstructed from sequences amplified by ORF1, RT, P1, and P4 primers were similar by resolving two groups, referred to as Clades I and II (Figure 3a, b, e, h). In contrast, the phylogeny reconstructed for Badna, ORF3A, P2, and P3 amplicons each resolved a previously unknown CSSV-like clade, referred to as Clade III (Figure 3 c, d, f, g).
The relationship among the isolates represented in each phylogenetic tree did not appear to be based solely on extant geographic origin. In all instances, Clade I contained isolates collected from central eastern and western Cote d’Ivoire, three GenBank reference sequences from Ghana (Accessions AJ608931, AJ609019, and AJ609020), and isolates from Togo(Accessions, AJ534983, AJ781003 and L14546). An exception to this pattern was observed by the ORF1 tree, where the three Togo reference genomes grouped in Clade II (Figure 3a). In contrast, for most trees, Clade II consisted primarily of isolates from eastern Cote d’Ivoire, GenBank Accession JN606110 from Cote d’Ivoire, and occasionally, the GenBank reference from Togo, Accession AJ781003, or R290 from western Cote d’Ivoire (Figure 3b, c). Overall, <33% of all amplicon sequences grouped in Clade II with the Cote d’Ivoire GenBank reference, even though all samples were collected in Cote d’Ivoire.
The nineteen amplicons obtained using the Badna, ORF3A, P2, and P3 primers, grouped in Clade III, sharing <80% nt identity with previously published CSSV sequences. These viral sequences were obtained from five of the 69 CSSV-positive samples, four of which were cacao while the fifth was X. maffafa (Figure 3 c, d, f, g). Neither the Badna amplicons (Clade III; Figure 3c), P2 (Figure 3f) nor the P3 primer pair-derived sequences (Figure 3g) grouped with apartial or complete genome sequence in GenBank. However, four ORF3A-amplicons clustered in Clade III with previously reported partial ORF3-amplicons from Cote d’Ivoire cacao samples [49]. Conversely, Clade I and II amplicons were most closely related to the ORF3A MP region, referred to as GroupA, B, and D, where as Clade III amplicons were closely related to groups E and F, per Kouakou et al. [49]; (Figure 3d). By comparison, none of the isolates identified herein clustered with Group A or C reference sequences, except for the R290MP1 isolate sequence that is positioned between these two groups in the phylogenetic tree.
In addition, by phylogenetic analysis, several amplicons from cacao and non-cacao hosts were basal to the three well-supported clades, indicating they diverged substantially from all other field isolates, thereby representing previously unreported CSSV-like types. Lastly, some amplicons were observed to ‘shift’ among clades I, II, and III, depending on the primers used for PCR amplification, and involved both cacao- and non-cacao samples. For example, R290 grouped in Clade I based on the RT, ORF3A, P2, P3, and P4-derived amplicons, in Clade II with respect to the RT-RNase H locus, and for the ORF1 amplicon, was basal to Clades I and II. Similarly, the Togo AccessionAJ781003 loci shifted between Clades I and II for ORF1, RT, RT-RNase H, ORF3A, and P1 amplicons, or were basal to the latter two clades, with respect to P2, P3 and P4 amplicons. Collectively, such amplicon-shifting is strongly suggestive of mixed infections and/or possibly, of interspecific recombination.
In this study, eight primers pairs evaluated for PCR-amplification of CSSV from DNA purified from cacao and non-cacao field-collected samples from Cote d’Ivoire, were shown to differ greatly with respect to amplification frequency. Amplification using the newly designed primer pairs, RT, P1, P2, P3, and P4, ranged from 23 - 42% (Table 2). By comparison with three previously reported primers, ORF3A, ORF1, and Badna [13,49,52] amplification of CSSV-like sequences was successful 19 – 33% of the time, making the newly designed primers somewhat more effective but not much more reliable. Results indicate that CSSV detection was dependent upon the viral region targeted by the particular primer pairs. Nevertheless, no one primer set amplified CSSV from all symptomatic samples despite amplification by at least one primer pair that confirmed positive infection. Although P4 primers had the highest amplification frequency, at 42%, they were unable to detect all of the isolates amplified by other primer sets that had overall lower frequencies, compared to P4. Although six of the eight primer pairs were degenerate and so, were expected to detect CSSV at a higher frequency than non-degenerate primers, they failed to meet this expectation. This was considered most likely due to the paucity of available information regarding true genomic variability among extant CSSV field isolates that precludes the consideration of global variability in primer design. Also, the intended use of the Badna primers was for genus-wide badnavirus amplification [52], however, when they were designed, only one CSSV genome sequence was available for consideration. This shortcoming in the Badna primer design, given the now apparently extensive divergence among CSSV isolates, appears to explain the basis for the lower than expected detection of CSSV e.g. in only 35 of 124 (28%) samples tested. Likewise, each primer pair gave ‘false negative’ results, based on the observation that 69 samples were confirmed positive for CSSV by at least one primer pair. Given the apparently greater than expected variability among CSSV-like isolate, the design and use of multiple instead of a single putative universal primer pair will probably be essential to develop a reliable molecular diagnostic test for CSSV.
Despite the characteristic CSSV-like symptoms, 55 of 124 field samples were negative for CSSV detection, regardless of primer pair. Primer design has been based on regions of high intra-isolate genomic conservation, and therefore was considered to represent informative regions for specieswide molecular detection. The lack of success to amplify virus from all symptomatic plants, despite a logical approach, indicates greater than expected genomic variability among CSSV-like field isolates, and may suggest that previously known genotypes have undergone differentiation, and/or that new genome types have emerged. Another explanation for the negative detection may be sample quality, low CSSV titer, or that symptoms observed in cacao, or the suspected wild hosts, are not caused by CSSV. Among the eight samples assessed for DNA quality by PCR amplification of the cacao nad 4 gene, results showed that all eight samples produced an amplicon of the expected size, indicating that the lack of amplification was not likely due to poor DNA quality. The results here in corroborate other failures to amplify CSSV by PCR amplification from multiple independently-collected field isolates [13,49] and further support the hypothesis that CSSV is more divergent than expected, an observation that has been reported for other badnaviruses, which exhibit inherent, intra-specific variability [68-70].
The overall genome sequence coverage made possible using the eight primer pairs accounted for 5 to 75% of an average-sized CSSV-like genome. Among the sequences, the variability was high, at 66 –99% pairwise nt identities. The locus with the greatest variability was that corresponding to the P3 amplicons, at 66-99% nt divergence, followed by ORF3A and Badna amplicon sequences, at 68-99% nt divergence, respectively. The P3 and ORF3A regions encode viral AP and MP, respectively, whereas, the Badna region encodes the partial RT and RNase H protein, collectively, the proteins involved in virus replication and in planta movement. Interestingly, the AP, MP, and RT-RNase H regions are about equally divergent, however, to what extent they have evolved compared to other genome regions among the isolates studied, is not yet known. Also, whether the AP and MP partial genome loci are similarly taxonomically informative as the RT-RNase H marker has not been evaluated.
The other five loci, RT, P1, P2, P4 and ORF1, which showed nucleotide variability at 71 – 99%, are less divergent than the AP, MP and RT-RNase H, and are therefore expected to readily detect the more conserved regions of the genome. Although the ORF3A, P3 and Badna loci are valuable at showing the extent of divergence of the CSSV genome, PCR amplification results showed that their amplification frequencies are not as high as that of the less divergent RT and P4 loci, for example (Table 2), which is expected. This suggests that the AP, MP and RT-RNase H may be going through more rapid evolutionary changes compared to all the other regions of the CSSV genome. That regions of virus genomes do not evolve at the same rate is consistent with a number of other plant viral groups.
Pairwise distance analysis of the 577 bp fragment of the taxonomically informative RT-RNase H region, which is delimited by the Badna primers, revealed the presence of four CSSV variants, based on the less than 80% nt identity threshold. Clearly, DNA sequencing of the entire RT-RNase H locus will be required to confirm species status, and corroborate the suggestion based on results herein that CSSV constitutes a complex of multiple, divergent entities.
The proposed extensive variability further explains the inability of most of the primer pairs evaluated here to amplify CSSV from all of the symptomatic plants analyzed in this study. Further, the apparently wide occurrence of single nucleotide polymorphisms in the viral genomes is expected to greatly limit or even preclude the design of “universal” primers for the group, albeit, variant-specific primer design may be tractable. Also, additional design attempts based on existing sequence data are expected to fail to amplify all isolates because the information regarding genomic variability is greatly limited. This extensive divergence suggests that variant-specific primers would offer the most likely-to-succeed strategy, however, far more CSSV genomic data are needed to test this hypothesis.
Phylogenetic analyses resolved a tree with two or three major clades containing sequences obtained as amplicons using the eight primer pairs. The sequences amplified by the ORF1, RT, P1, and P4 primers were grouped into two major Clades, I and II (Figure 3a, b, e, and h). In contrast, the trees based on the sequences amplified by the ORF3A, P2, P3, and Badna primers showed an additional third clade, III, which grouped separately, basal to the Clades I and II (Figure 3 c,d, f,g). The ORF3A and P3 regions appear to be more divergent than the other six regions because they resolve the tree sub-structure better as an indicator of genomic variability. They both resolved six or more CSSV subgroups of isolates in samples from Cote d’Ivoire and Ghana [49,50] (Figure 3d, 3g). Among them, ORF3A sequence subclades are the best resolved, most likely because there are more previously published partial sequences available for this region [49] included in the analysis, while no partial fragments corresponding to the P3 region are available, except for the full-length genomes. For the same reason, the badnavirus-taxonomically informative RT-RNase region [19] shows less genomic variability, given that only a few sequences were considered in the phylogenetic analysis, compared to MP and P3 regions. Such high variability was also observed with the nucleotide pairwise analyses, and may be attributed to a high rate of mutation or evolution. Although some loci had more sequences than the P3 and the ORF3A considered for the phylogenetic analyses, for example, the RT and P4 regions, they showed less genomic variability because they resolved less subgroups. In summary, the results suggest that some CSSV loci are better at resolving genetic variability, while some are better at detecting more CSSV isolates.
Phylogenetic analyses based on the ML method showed no evidence of phylogeographical distribution for the sequences analyzed here. At least two-thirds of the sequences in this study from Cote d’Ivoire represented across the eight phylogenetic trees were most closely related to the Ghana GenBank reference sequences in Clade I (Figure 3). Clades II and III contained GenBank reference sequences from Togo and/or Cote d’Ivoire, or none. This distribution suggests that the CSSV isolates studied here may not be endemic to Cote d’Ivoire, and that representatives of each group of isolates may be present in all cacao-growing countries in West Africa. Also, within, Cote d’Ivoire, there was no evidence for close relationships between CSSV isolates based on sampling location or plant host species, in that eastern, western, or central region isolates from cacao and non-cacao hosts were equally distributed among the clades. Because of the exchange of germplasm among cacao growers in the cacao-growing regions in West Africa, and also movement of planting material in search of new locations that are not affected by the swollen shoot disease, it is expected that several isolates or variants of CSSV be present in the region.
The Badna, ORF3A, and P3 amplicons that grouped within Clade III also shared the least nt identity with other isolates by pairwise distance analysis. And for the Clade III taxa, no close relative (≥ 80% identity) was identified among the CSSV sequences in GenBank, indicating they represented highly divergent isolates.
CSSV sequences were not detected only in the cacao host, they were also amplified by all primers but P1 and ORF1, from seven wild host species; C. papaya, C. pentandra, C. erecta, D. cayenensis, S. anthelmia, T. bangwensis, and X. maffafa. Of the seven, C. pentandra has been previously described as an alternative host [15]. The CSSV sequences from the seven wild host plants and the cacao samples from Cote d’Ivoire grouped in the same subclade with Togo, Cote d’Ivoire, and Ghana isolates (Figure 3b, c, d, g, and h), indicating a close evolutionary relationship. This supports findings from previous studies that wild hosts serve at least in part as the source of CSSV, and it is well known that CSSV was present in West Africa in wild hosts prior to the introduction of cacao in the 1800’s [14,15]. Results suggest that use of cover crops in cacao plantations that have not been evaluated with respect to CSSV susceptibility may harbor CSSV and lead to unintentional cultivation of an alternative host from which the virus can spread into the cacao trees they are intended to protect. Also, establishing new fields near native forests may periodically lead to host shifting by CSSV or other as yet uncharacterized endemic badnavirus species into newly planted cacao trees.
The five primer pairs designed here in for CSSV detection, collectively, amplified badnavirus amplicons from 19-42% of the cacao and noncacao field samples collected in Cote d’Ivoire. Based on the collective sequence coverage (AP, MP, RT-RNase H, RT, P1, P2, P4 and ORF1) of the CSSV-like amplicons, enabled by amplification of eight primers in total (including positive control primers), unexpectedly extensive variability was uncovered, at 66 – 99% nt identity. Given the current ICTV species cut-off at ≥80%, for the RT-RNase region, and the amplification herein of a 577 bp fragment of that locus (~47%), it is not possible to determine if this extent of genomic variability (representing 5 -75% of the total genome) is a valid indicator of species divergence, however, results provide important new clues that support this possibility, and one that implicates CSSV as complex of at least four or more variants. This provisional scenariois consistent with the observed similarly high divergence among badnavirus Banana streak virus genomes [68], and regarding certain emergent begomoviral species groups in the family Geminiviridae [71]. Given these results, it is unlikely that a single PCR primer pair can be designed for detection of all extant CSSV-like or other badnaviral variants. This underscores the urgency to develop new molecular or other diagnostic tools that enable reliable detection and identification of cacao-infecting badnaviruses responsible for unprecedented losses in cacao plantations throughout West Africa. Comprehensive molecular diagnostic tests would be invaluable for confirming the virus-free status of cacao clones destined for new production areas and for tree replacement programs in extant cacao-growing locales, as well as to provide support ongoing breeding efforts to identify CSSV-resistant germplasm. In addition, the application of next-generation genomic pathology approaches to epidemiological studies will be invaluable for reconciling the extensive genomic variability occurring among suspect new and emerging badnaviral species of cacao, with viral evolutionary patterns in the context of the current pandemic spread, and with respect to comprehensive cacao genetics/genomics data sets, to achieve long-term disease management solutions to enable sustainable cacao production in West Africa.
The authors declare no conflict of interest.
Funding for this project was provided by USDA-ARS through a Specific Cooperative Agreement, Project # 6038-21000-023-07, Development and Optimization of Molecular Diagnostics Method for Qualitative and Quantitative Detection of Cacao Swollen Shoot Virus, and from MARS, Inc.; Trust Agreement #58-6631-6-123: Genetic Improvement of Theobroma cacao, and by the USDA-Foreign Agricultural Services, World Cocoa Foundation, Borlaug Fellowship Program that was awarded to the second and third authors.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation:Chingandu N, Kouakou K, Aka R, Gutierrez OA, Brown JK (2017) P Unexpected Genome Variability at Multiple Loci Suggests Cacao Swollen Shoot Virus Comprises Multiple, Divergent Molecular Variants. J Emerg Virol Dis 3(1): doi http://dx.doi.org/10.16966/2473-1846.128
Copyright: © 2017 Chingandu N et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access