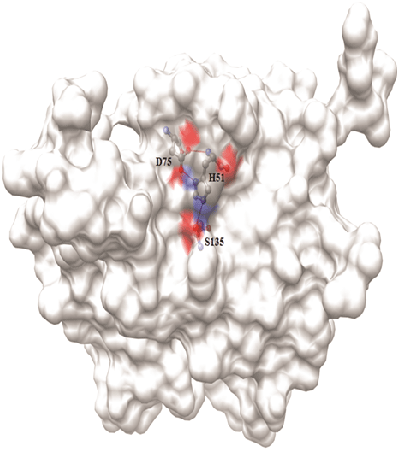
Figure 1: Surface diagram of Den2 NS2B/NS3 protease with its catalytic triad
NHV Kutumbarao1 Chandrasekaran Ramakrishnan2 Krishnamoorthy Balasubramanian3 Devadasan Velmurugan1,*
1Centre of Advanced Study in Crystallography and Biophysics, University of Madras, Maraimalai Campus, Guindy, Chennai, India*Corresponding author: D Velmurugan, UGC-BSR Faculty, CAS in Crystallography and Biophysics, University of Madras, Guindy Campus, Chennai, Tel: 9841075847; E-mail: shirai2011@gmail.com
NS2B/NS3 protease complex plays an essential role in replication of dengue virus (DENV) in the host cell. NS3 protease is a trypsin-like serine protease and NS2B acts as a cofactor. The active site of NS3 protease contains a catalytic triad made up of His51, Asp75, and Ser135 at the N-terminal region and it is involved in cleavage of the poly-protein into single functional units. Hence, targeting NS3 protease is more important in development of more potent inhibitors to control the propagation of DENV. In this study, mode of binding of acridone and xanthones, the cyclic ketones are studied with reference to that of the experimentally proven flavones and chalcones. Docking results suggest that acridone and xanthone prefer to bind the new site (the tunnel like pocket) whereas the flavone based inhibitors and cardamonin prefer to bind the active site with the catalytic triad. Moreover, the results lead to postulate the synergistic activity of xanthone/acridone with flavone derivatives to control the DENV infection. Molecular dynamics simulation and binding free energy calculations confirm the stability of acridone and flavone which bind at the tunnel and catalytic sites, respectively.
Dengue virus; Acridone; Xanthone; Flavone; NS2B/NS3 protease; Docking; Flavivirus
Dengue virus (DENV) belongs to the Flavivirus genus and Flaviviridea family that comprises four unique serotypes viz., DEN-1 to DEN-4. It causes dengue hemorrhagic fever, an infectious disease in most of the tropical countries. Rapid onset of fever, joint and muscle pains, headache and appearance of rashes are the immediate symptoms upon infection [1,2]. Transmission of DENV is particularly by Aedes aegypti, a mosquito that commonly spread the viruses responsible for dengue fever, chikungunya, yellow fever, etc in tropical and subtropical regions. Dengue virus type 2 (Den2) is the common one of the four serotypes and its genome is a stretch of 10,732 nucleotides in the form of single-stranded positive-sense RNA. It encodes a poly protein precursor of 3,391 amino acids in length [3,4]. It is organized in the form of C-prM-E-NS1-NS2ANS2B-NS3-NS4A-NS4B-NS5 including three structural proteins (C, prM and E) and seven non-structural proteins (NS) [5,6]. The discussions on the functional aspects of the Dengue NS2B/NS3 protease, its structural properties, drug design strategies, and resulting inhibitors are available in the literature [7-9]. In addition, the crystal structure of NS3 protease domain in complex with the 47 residue fragment of NS2B revealed the key residues involved in substrate recognition and mechanism of activation of NS3 [10] . For proteolytic cleavage of the poly-protein, the NS3 protease needs ~ 40 residue hydrophilic domain from NS2B [11]. Moreover, His51, Asp75 and Ser135, the active site residues form the catalytic triad (Figure 1) are responsible for the proteolytic activity which is the exclusive mechanism of action of serine protease family [12,13]. Lys74, Leu149 and Asn152 are located near the active site and interact with the inhibitors through hydrophobic contacts. Binding of the inhibitors induces conformational changes in the active site of NS2B/NS3pro and breaks the H-bond interaction between the Asp75 and Lys74. The NS2B/NS3 protease complex is taken as the target for the present study for development of anti-dengue compounds [14]. Due to endemic, rate of development of inhibitors of NS2B/NS3 protease has increased. Discussion on methods of development [15,16] of some of these inhibitors is more relevant to the present work. It includes the peptido-mimetics synthesized based on the substrate peptide [17-19] or cyclo-peptides [20]. Structure-based virtual screening method was efficiently used for finding novel inhibitors [21-23]. Especially, the anthrecene based compounds ARDP0006 and ARDP0009 are structurally similar to acridone and xanthone and show considerable inhibitory activity in cell based replication assay at micro molar concentrations [22]. Similarly, library of natural products [24-26] extracted mainly from Boesenbergia rotunda (L.) Mansf. Kulturpfl (BRI) and a common spice of the ginger family (Zingiberaceae) is the source for flavone based compounds reported as inhibitors of NS2B/NS3 protease. Small molecule libraries [27] were screened to identify NS2B/NS3 specific inhibitors by in vitro high throughput screening (HTS) method and the compound BP2190 is reported as a novel and selective inhibitor [27]. Virtual screening and core hopping techniques together short listed some of the non-peptide inhibitors from ACD chemical library [28]. Nevertheless, only less number of inhibitors including peptide or small molecule inhibitors with moderate activity has been reported. Benz[d] isothiazol-3(2H)-one derivatives made by click chemistry were proven as anti-viral compounds against Den2 and West Nile virus (WNV) [29]. Derivatives of anthracene show inhibitory activity at micro molar concentrations which was proven by protease inhibitor assays and structure activity relationship (SAR) study [22]. The RC-1 peptide inhibits the NS2B-NS3pro with the IC50 value of 46.1 µM at 28°C, 21.4 µM at 37°C and 14.1 µM at 40°C [30]. Protegrin-1 (PG-1), a 18 a.a. peptide has been reported as a Den2 protease inhibitor by MK2 cell line based assays. It also controls the viral replication in the host and it shows the toxicity at the concentration of 12.5 µM and above [31]. Non-substrate based NS3 protease inhibitors were also reported using homology modeling, virtual screening and docking. However, there are no crystal structures available for the complex form of Den2, the NS2B/NS3pro complex with its inhibitors.
Figure 1: Surface diagram of Den2 NS2B/NS3 protease with its catalytic triad
Boesenbergia rotunda L, contain a set of compounds including flavones and their corresponding chalcones was reported as good noncompetitive inhibitors of Den2 protease [24]. Flavones (pinostrobin, pinocembrin and alpinetin) and their chalcones (pinostrobin chalcone, pinocembrin chalcone and cardamonin) were found to bind at the active site by the rigid and automated flexible docking methods with a good correlation between the predicted and experimental binding affinity [32]. Availability of data that support antiviral activity of some of the flavones and their chalcones are highly helpful for analysis of structurally similar compounds such as acridones and xanthones. Moreover, acridones are proven as antiviral compounds as they inhibit replication of herpes simplex virus (HSV) [33] and RNA synthesis in Junin virus [34]. Most importantly, activity of N-substituted acridones against haemorrhagic fever viruses like dengue were also been reported [35]. A recently published work reports the potential role of acridone and their derivatives in inhibiting the Dengue viral replication. The inhibition is found towards all the four serotypes, with increased inhibition towards DENV-2. The mode of inhibition has been studied with a host enzyme called inosinemonophospate dehydrogenase. This enzyme is believed to have an effect on viral replication by restricting the GTP synthesis [36]. The potential of acridone for anti-viral activity by viral protease inhibition and its increased efficiency by dual mode of binding is undertaken in the present study. On the other hand, xanthones are very specific group of biologically active compounds. Many of its derivatives are known to have a strong antiviral activity [37] and 20 different xanthones isolated from Swertia mussotii are known to inhibit viral replication in hepatitis B virus (HBV) infected cells [38]. In addition, mangosteen (Garcinia mangostana) fruit is one of the natural sources of many bioactive xanthones and it shows good antiviral and anticancer activities without adverse side effects. In general, both acridones and xanthones are common in terms of their inhibitory activity against the different types of Herpes Simplex Virus (HSV) [39]. Hence, the present study aims at the possibilities for inhibition of NS2B/ NS3pro complex by acridone and xanthone derivatives with reference to the already reported flavone based inhibitors.
The crystal structure of NS3 protease in complex with the fragment of NS2B cofactor was taken from the Protein Data Bank (PDB ID: 2FOM) [10]. In the structure, a loop region is missing, which has been modelled using PRIME available in Schrödinger. The protein was prepared by assigning proper bond order and formal atomic charges. Proper ionization and tautomeric forms were ensured and optimized the H-bond network. A regular protocol of “protein preparation wizard” was used to sample the conformations of hydroxyl and thiol groups, amide group of Asn and imidazole group of His. All the amino acids (especially, Asp, Ser, Glu, Arg and His) were protonated. Before starting the docking calculation, the whole structure of the NS2B/NS3 protease was optimized and minimized with Root Mean Square Deviation (RMSD) cut off of 0.3Å in order to remove steric clashes and to calculate the conformation corresponding to (nearest) local minima.
Flavones (pinostrobin, pinocembrin and alpinetin) and a chalcone (cardamonin) are taken as standard (known inhibitors of Den2 NS2B/ NS3 protease) and their structures are depicted in figure 2. The derivatives of acridone and xanthone (Table 1) were drawn and optimized using the Chemsketch software. All these compounds were prepared for docking using “Ligprep” module available with Schrodinger Suite. The default parameters were set to generate maximum of 32 tautomers and stereoisomers at pH 7 ± 2. Steepest descent followed by conjugate gradient methods was used for energy minimisation which is available under the Impact module for convergence of structure, geometrically as well as energetically.
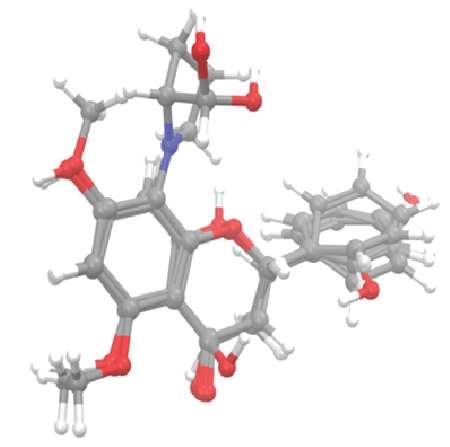
Figure 2: Alignment of 3D structure of flavone derivatives
The compounds already reported based on experimental activity study were docked using induced fit docking (IFD) method [40]. It produced multiple binding poses for each ligand and ranked primarily by docking score and calculated glide energy. In this method, both the receptor and the ligand are allowed to move. Hence, the binding site can adopt the conformation that is suitable for the ligand and vice versa. The best pose for each standard ligand was chosen based on score, energy and the interactions. In order to find the site preference for xanthone, acridone and flavone, blind docking was performed using Glide [41] package. During the process, the grid was set to the whole protein to allow the ligands to find their binding pocket that is energetically favourable. Since the acridone and xanthone derivatives bind at the site adjacent to the catalytic site (aka, tunnel site), the IFD method was employed to assess the synergistic inhibition of Den2 protease by acridone and xanthone while flavones bind at catalytic site. The interactions between protein and ligand were analyzed using Ligplot [42] and Chimera [43] software packages. Optimized Potential for Liquid Simulation (OPLS-2005) [44] force field was used for all the above calculations. MD simulations were performed to confirm the stability of acridone and xanthone binding at the tunnel site while flavone binds at the catalytic site.
NS2B/NS3 protease in complex with each of the best scored inhibitors was simulated for 10 ns [45]. Simulation system for each protein-ligand complex was prepared using Leap module available with AMBER10 suite [46]. The established procedure was adapted to impart protonation states of the amino acids. For charge neutralization of the system, Na+ or Clwas used. The force field parameters for acridone, xanthone and flavone derivatives were derived using antechamber program and GAFF (general amber force field) [47]. AM1-BCC charge method was used in assigning the atomic point charges [48]. The TIP3P [49] box (triangulated water molecules) with 10 Å3 box was invoked for solvation. Steepest Descent (SD) and Conjugate Gradient (CG) methods were employed to minimize the system in two phases. In the first phase, solute atoms were restrained by harmonic potential with force constant (kcal/mol-Å2 ) and the solvent atoms were relaxed. Then, CG method was employed to relax the whole system without restraining the solute atoms. In order to set the system to obey NPT ensemble during simulation, equilibration process was carried out in two phases. First, the temperature was equilibrated around 300 K using Berendsen temperature coupling [50], in which, 2 ps time constant was set to temperature coupling and the Maxwell distribution was used to generate random seed which was used to assign initial velocities for given temperature.
The periodic boundary condition (PBC) was implemented along with constant volume. In the second stage, isotropic positional scaling with time constant (2 ps) for pressure coupling were set to equilibrate the pressure around 1 atm. Velocity information was taken from the end of previous equilibration stage. After the equilibration, the system was interpreted in terms of the potential, kinetic and total energy as indicators to analyse the finished equilibration steps. Finally, 10 ns simulation was performed with 2 fs integration time without solute atoms being restrained. The ff03 [51,52] all atom force field was used in all the steps. Both the equilibration and MD simulations were carried out using Particle Mesh Ewald (PME) method [53] to treat the long range electrostatic interactions, SHAKE algorithm [54] to restrain all the bonds involving hydrogen atom (to control fastest vibrations), 2 fs time step and 10 Å non-bonded interaction cut-off.
The binding free energies (ΔG°) were calculated for complex formation between NS2B/NS3 protease and acridone or xanthone binding at tunnel site. MM-GB/SA [55-57] method and MMPBSA.py [58] module were employed for calculations involving the last 2 ns (with 5 ps frequency) of 10 ns trajectory. In MM-GB/SA method, the ff03 force field was used to calculate the MM energy (eq.2). GB method [59] was used to calculate the polar solvation energy and the molsurf [60] was used to calculate the solvent-accessible surface area (SASA). The nMode module was used by MMPBSA.py program to estimate the entropic contribution (-TΔS). The overall calculation is described in the following equations.
\[\Delta {G^o}_{bind,solv} = \Delta {G^o}_{bind,vacum} + \Delta {G^o}_{solv,complex} - (\Delta {G^o}_{solv,ligand} + \Delta {G^o}_{solv,receptor})\,\,\,\,\,\,\,\,\,\,\,(1)\]
\[\Delta {G_{bind}} = \Delta H - T\Delta S \approx \Delta {E_{MM}} + \Delta {G_{sol}} - T\Delta S\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,(2)\]
\[\Delta {E_{MM}} = \Delta {E_{{\mathop{\rm int}} ernal}} + \Delta {E_{electrostatic}} + \Delta {E_{vdw}}\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,(3)\]
\[\Delta {G_{sol}} = \Delta {G_{PB/GB}} + \Delta {G_{SA}}\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,(4)\]
Where, ∆EMM is gas phase molecular mechanics energy; − T∆S is conformational entropy; ∆Gsol is solvation free energy; ∆Einternal includes bond, angle and dihedral energies.
As a result of IFD, 20 binding poses for each ligand were produced and the score and energy for each pose was obtained. Score of -6 ± 1 kcal/ mol was observed for all the standard compounds (cardamonin, alpentin, pinocembrin and pinostrobin) (Table 1). These compounds bind well at the catalytic site and interact with all the three residues of catalytic triad and other neighboring residues through hydrogen bond and hydrophobic interactions. Score and binding energy are in comparable range as well as in the range of the values already reported in the literature [32].
Compounds |
Binding Site |
Score |
Energy (kcal/mol) |
Cardamonin |
Catalytic triad |
-6.19 |
-33.87 |
Alpenetin |
Catalytic triad |
-5.34 |
-32.91 |
Pinostrobin |
Catalytic triad |
-5.70 |
-29.42 |
Pinocembrin |
Catalytic triad |
-6.57 |
-35.90 |
Acridone |
Nearby the Catalytic triad |
-7.83 |
-53.12 |
Xanthone |
Nearby the Catalytic triad |
-7.92 |
-50.19 |
Table 1: Score and energy of each compound docked with NS2B/NS3 protease
Further, the nucleus structure of acridone and xanthone were subjected for Glide docking without specifying the binding site (blind-docking). Table 1 and figure 3 detail the score, energy and interactions associated with these compounds binding at the site nearby the catalytic triad (tunnel site). Both the acridone and xanthone prefer the tunnel site with score -7.8 kcal/mol which is better than that is observed for binding of standard compounds at the catalytic triad. Important characteristics of binding at the tunnel include interaction with the Lys73 and Lys74. The role of these two lysine residues is very important for binding of ligands at the catalytic triad. Binding of acridone and xanthone at the tunnel through their non-bonded interactions with Lys73 may induce conformational change at the junction of both the sites. The tunnel site is mainly composed of many hydrophobic (mainly, Lys73, Lys74 and Lue76) and few polar (mainly, Thr120 and Asn167) amino acids. This hydrophobic pocket possesses more void volume even in the presence of acridone or xanthone as shown in surface diagram in figure 3 (right panel). In order to improve the binding efficiency, different derivatives of acridone and xanthone are modelled by substituting different functional groups rich of hydrophobicity. Methyl, isopropyl and tert-butyl groups were substituted at X1 and X6 positions which are expected to improve the hydrophobicity as well as shape complementarity with the tunnel site. The details of substitutions made in both the core structures are described in table 2. The 16 derivatives for xanthone and acridone, separately, are subjected for IFD to find the favourable substitutions with good score and energy. IFD results confirm that the isopropyl group substituted at both X1 and X6 positions of both acridone and xanthone improved the binding efficiency (binding energies: -50.1 and -53.1 kcal/mole, respectively) with the interaction pattern similar to their respective core structures. Xanthone derivative has favourable score of -8 kcal/mol whereas it is -7.8 for the acridone derivative (Figure 4).
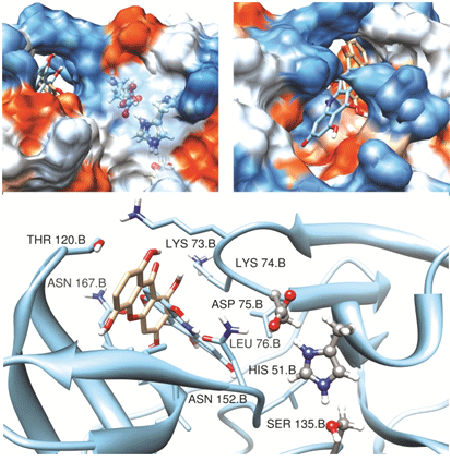
Figure 3: Binding of xanthones and acridones at the tunnel site. The surface diagrams depict catalytic triad residues (ball and stick, left panel) and the tunnel site together and full view of tunnel site with the ligands (right panel). The same is depicted using cartoon to understand the binding residues from both the sites
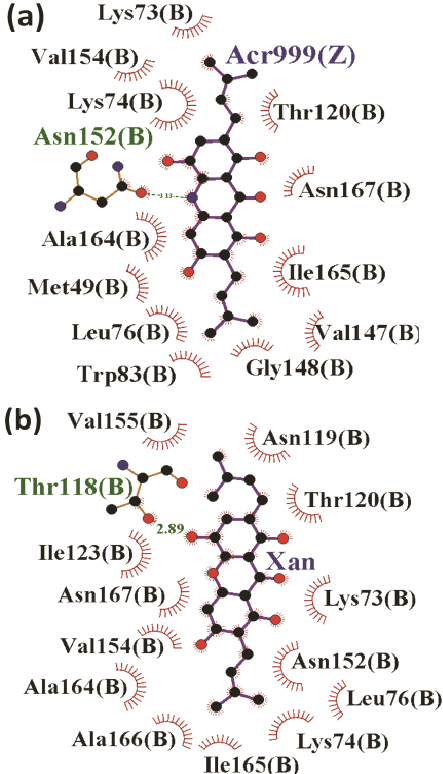
Figure 4: Binding mode of acridone (a) and xanthone (b) at the tunnel site of Den2 protease
|
||||||
Sl. No. |
X1 |
X2 |
X3 |
X4 |
X5 |
X6 |
1. |
H |
H |
H |
H |
H |
H |
2. |
Me |
H |
H |
H |
H |
Me |
3. |
iP |
H |
H |
H |
H |
iP |
4. |
tBu |
H |
H |
H |
H |
tBu |
5. |
H |
H |
H |
Me |
H |
H |
6. |
Me |
H |
H |
Me |
H |
Me |
7. |
iP |
H |
H |
Me |
H |
iP |
8. |
tBu |
H |
H |
Me |
H |
tBu |
9. |
H |
H |
H |
Me |
H |
H |
10. |
Me |
H |
H |
Me |
H |
Me |
11. |
iP |
H |
H |
Me |
H |
iP |
12. |
tBu |
H |
H |
Me |
H |
tBu |
13. |
H |
H |
H |
H |
H |
H |
14. |
Me |
H |
H |
H |
H |
Me |
15. |
iP |
H |
H |
H |
H |
iP |
16. |
tBu |
H |
H |
H |
H |
tBu |
Table 2: Derivatives made by substituting the selective functional groups in the different positions of core structure of acridone and xanthone
Functional groups: H-Hydrogen atom; Me- methyl; iP-isopropyl and tBu- tert-butyl.
Further, docking was performed to assess the binding efficiency of each flavone derivative and the cardamonine at the catalytic triad in the presence of acridone and xanthone at the tunnel site. The xanthone derivative binds to the tunnel site with more number of hydrogen bond interactions than that of the acridone derivatives. The xanthone derivatives interact with Lys73, the hydrophobic amino acid present in the junction of the tunnel and the catalytic sites. In addition, these derivatives interact with Val155 and Ala166 present in the tunnel site. Apart from the hydrogen bond interaction, hydrophobic contacts with the other amino acids increase the stability of the complex. On the other hand, acridone derivatives interact with Leu149 and Asn152 through hydrogen bond as well as hydrophobic interactions
The above results confirm that flavones and cardamonine prefer the catalytic-triad region whereas the xanthone and acridone derivatives prefer to bind at the tunnel site. Hence, the docking was performed for all the standard compounds at the catalytic triad in the presence of xanthone and acridone at tunnel site, separately. The results will be much helpful to identify the different combinations of these two groups of compounds binding at their respective sites, hence to prove the synergistic function of ligands. In the first step, the xanthone was docked at the tunnel site and screening of the standard compounds was carried out at the active site. It confirmed that the flavone binds well with the best glide energy of about -69.9 kcal/mol . Similarly, the acridone derivative was fixed at the tunnel region and the screening was carried out at the triad where acridone derivative shows good binding energy (-53.1 kcal/mol) (Figure 5a). In the presence of acridone at tunnel site, binding energy for flavone to bind the active site (catalytic triad) is -40.7 kcal/mol (Figure 5b). This is highly unfavourable when compared to its binding in the presence of xanthone (in the place of acridone) (Figure 5c). The binding free energy calculated (entropy term included) based on the MD simulation trajectory substantiates well the above docking results (Figure 6). Acridone binds very strongly (-24 kcal/mol) compared to the xanthone (-6 kcal/mol). Binding of second acridone (at catalytic triad) is not favourable (< -3kcal/ mol) whereas flavone binds well (-4 kcal/mol). Similarly, flavone shows more favourable binding (-7.5 kcal/mol) in the presence of xanthone at the tunnel site. During synergistic inhibition with flavone, acridone binds tunnel site with more affinity (-12.5 kcal/mol) compared to the xanthone (<-3 kcal/mol). Together, acridone and flavone show favourable binding with their preferred sites suggesting that these two compounds will have synergistic inhibitory activity against the proteolytic activity of NS2B/NS3pro.
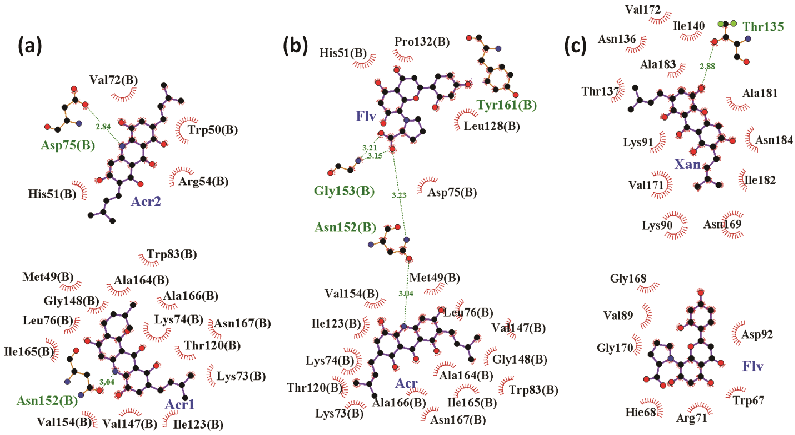
Figure 5: Synergistic binding mode of acridone and xanthone at tunnel site while flavone/acridone binds in catalytic site
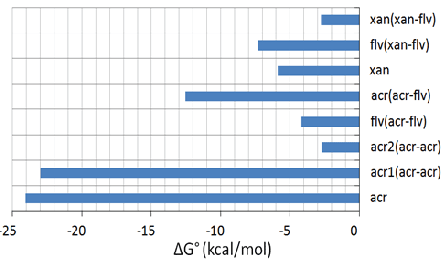
Figure 6: Binding free energy (ΔG°=ΔH-TΔS) calculated for binding of acridone and xanthone at the tunnel site in the presence of flavone at catalytic site. In acr#(acr-acr), possibility for acridone to bind both the sites, where, acr1 denotes binding of acridone at the catalytic site and acr2 denotes binding of acridone at the tunnel site
Flexible docking studies confirm that the flavone and chalcone based inhibitors of DENV NS2B/NS3pro prefer to bind the active site with the catalytic triad. The acridone and xanthone based compounds are known to have antiviral activity and they tend to bind at the new site nearby the catalytic triad, called tunnel site. This provides a new insight in designing inhibitors for DENV based on the structure of NS2B/NS3 protease. Also, it provides a base to propose that a synergism can exist in the inhibitory activity of these two site specific inhibitors. Flexible docking , MD simulation and binding free energy calculations confirm that acridone and flavone based compounds will synergistically inhibit DENV NS2B/NS3 protease complex and this new finding provides the base for further extension with biochemical and structural studies to confirm the same. Binding studies for the dual inhibition sites have not been reported so far. As a continuation of the present work, binding studies with anti-viral compounds from natural resources are being undertaken with NS2B/NS3 protease.
The authors thank Department of Biotechnology and University Grants Commission, Government of India for commercial software packages and high performance computation facilities. DV thanks DBT for funding the international collaborative project with Leubeck University.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation: Kutumbarao NHV, Ramakrishnan C, Balasubramanian K, Velmurugan D (2016) Computational Assessment of Inhibitory Activity of Acridone, Xanthone and Flavone Derivatives against NS2B/NS3pro of Dengue Virus Type 2. J Emerg Dis Virol 2(4): doi http://dx.doi.org/10.16966/2473-1846.124
Copyright: © 2016 Kutumbarao NHV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access
