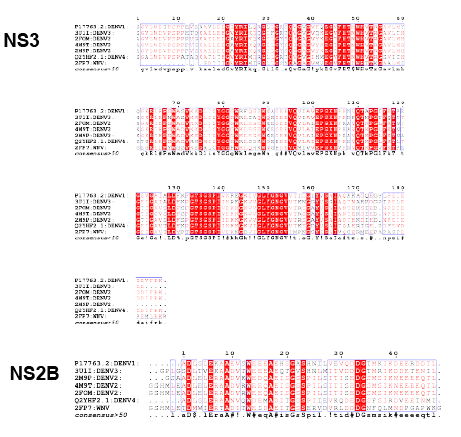
Figure 1: The sequence comparison of the protease NS3 and NS2B
NHV Kutumbarao D Velmurugan*
CAS in Crystallography and Biophysics, University of Madras, Guindy Campus, Chennai, India*Corresponding author: D Velmurugan, UGC-BSR Faculty, CAS in Crystallography and Biophysics, University of Madras, Guindy Campus, Chennai, India, Tel: 9841075847; E-mail: shirai2011@gmail.com
Dengue is one of the life threatening diseases. The non-structural protein3 (NS3) is the virus protease essential for the polyprotein processing which requires the presence of ~40 residue hydrophilic domain from NS2B cofactor for its optimal catalytic activity. The complex of NS2B/NS3 active protease is categorized as a trypsin like protease. Design of peptide based drugs towards the protease is one of the widely accepted strategies against this virus. The conformation adopted by NS2B plays an important role in the activity of the protease and also in the binding of the substrate. We have attempted to design peptides against NS2B/NS3 through various approaches. The ligands were selected from different sources, from peptides isolated from edible fishes to natural compounds isolated from papaya leaves. The binding mode of the ligands was studied with respect to different conformations of the protease.
DENV; Induced fit docking; Glide; OPLS force field
DENV: Dengue Virus; WHO: World Health Organization; OPLS: Optimized Potentials for Liquid Simulations.
Dengue virus (DENV) belongs to Flavivirus. Four antigenetically related serotypes are present, namely DEN-1, DEN-2, DEN-3, DEN-4. The virus is transmitted by Steomiya aegypti (Aedes). All the four serotypes are responsible for the invocation of the hemorrhagic fever [1]. The WHO has categorized Dengue as the most important mosquito-borne tropical disease. The symptoms of dengue infections vary from flu-like illness (dengue fever) to dengue shock syndrome and in cases the most severe dengue hemorrhagic fever (severe dengue with bleeding abnormalities). Dengue hemorrhage diseases are life-threatening.
Dengue virus genome is a single-stranded positive-sense RNA [2,3] that consists of 10,723 nucleotides which encodes a single polyprotein precursor which constitutes two categories of proteins, structural and nonstructural proteins. There are three structural proteins (C, prM and E) and a lipid bilayer around the RNA genome [4], where the protein C (core nucleocaspid protein) binds to RNA directly and the protein E (major envelope protein) and protein M (membrane protein) both form the protein outer shell [5]. Seven non-structural proteins (NS) are present, namely, NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5. The polyprotein precursor undergoes cleavage (co- and post-translationally) to produce mature proteins. The NS3 responsible for this activity, makes the virus active and helps in further replication. The NS3 acts specifically at region for the cleavage activity, the regions are NS2A/NS2B, NS2B/NS3, NS3/ NS4A and NS4B/NS5 at the nonstructural protein region [6-10].
The importance of the protease activity in the viral survival and replication and the notion of protease inhibitors being commonly viewed as a potential drug in many cases [11-13] led the research community to design active inhibitors to tackle the dengue infection. The protease domain NS3 fragment which is 180 amino acid in length is present on the N-terminal of the total 618 residue length multi domain NS3 [14- 20]. NS2B (cofactor) is essential for the activation of the NS3 protease activity [16]. The binding of the NS2B might initiate a structural arrangement of the active catalytic triad for the optimal protease activity [21,22]. The protease has specificity for substrate binding, which has been demonstrated by many workers. The results of Niyomrattanakit concludes that the preference for P1 and P2 positions are dibasic residues, with basic or aliphatic residues at P3 and P4 and P1’ with smaller or polar residues [23]. The minimal length of the NS3 determines the protease activity. The 47 residue length of the NS2B is determined essential for the protease to be active. The glycine linker connected NS2B to NS3 is soluble and enzymatically active [19,24]. The protease sequences have high sequence similarity within the serotype, the active site residues are conserved over all the serotypes. The sequence alignment of the different protease structures solved from two different serotypes has been represented in the figure 1 [25]. The high similarity between the sequences is evident. This can be observed even in the case when compared with west Nile virus protease also. The DEN2 and DEN3 serotypes proteases can be observed as highly conserved, especially around the active site residues, whose color is in red. The NS2B region too has the similarity between the stereotypes which allows us to model the missing segment of one protease serotype from the other.
Figure 1: The sequence comparison of the protease NS3 and NS2B
The NS2B/NS3 protease is a typical serine protease and the first NS2B/ NS3 crystal structure was solved from DEN2 strain at 1.5 Å resolution (PDBID: 2FOM) [26] This is a beta barrel conformation which is similar to chymotrypsin which has active site composed of three major residues, Histidine (HIS), Aspartic acid (ASP) and Serine (SER), which is named as catalytic triad. This structure has a gap in the loop region of the NS2B. Many of the subsequent structures solved have missing loops, one structure solved from DEN2 (PDBID: 4M9T) [27] with reported allosteric site has the trace of the loop and the orientation of the NS2B was similar to that of 2FOM structure. But a solution structure (PDBID:2M9P) recently deposited in PDB has an inhibitor bound in the active site and the NS2B region shows a major conformational change which was similar to the conformation of protease (PDBID:3U1I) solved from DEN3 [28] .The 2FOM structure which was the earliest solved structure has been recently reported to be in the inactive conformation. The orientation of the NS2B fragment in this conformation is compared to that of the structure 4M9T which was a mutant structure for A125C, and this structure is reported to have helped in the identification of the allosteric region (ALA 125) in the protease. The movement of the loop is identified to have an influential role. The conformations of the loop120 (117-122) and loop150 (153−164) [27] are crucial and influence the orientation of the NS2B fragment. The different positions of the loop with respect to the NS2B orientation and also the place of the ligand can be seen in figure 2. The movement of these loops is indeed linked to the binding mode of the ligand. This can be seen from the binding of different ligands shown in figure 2. The RMSD (Root Mean Square deviation) and orientations of superimposed structure 2FOM with 3U1I is 0.6 Å (127 atom pairs included) where as there is a deviation of 1.03 Å with 2M9P (only 80 atom pairs included). The RMSD between 3U1I and 2M9P is 1.18 Å (88 atom pairs included). There is a conformational similarity between 3U1I (DEN3) and the protease from West Nile virus, 2FP7 [26]. The positioning of the loop 120 is further deviated in the recent solution structure and the NS2B also. This can be inferred as the effect brought by different binding modes of the ligands. The various superimpositions of the structures are presented in figure 2. As the conformation adopted by the protease in 2FOM structure is inactive [29], many workers have used the subsequent structure such as 3U1I from DEN3 for modeling studies. Molecular docking and simulation studies were undertaken to understand the different binding modes of the ligands and the effect of placement of the loops for the ligand binding. The previous modeling studies were carried out using 2FOM as the target and we have analyzed the recent structures deposited and carried out the modeling analysis which lead to the binding mode of the few peptides similar to the binding mode of ligand seen in the 2M9P solution structure. The structure used for modeling from NMR (Nuclear Magnetic Resonance spectroscopy) studies is the one with the least energy.
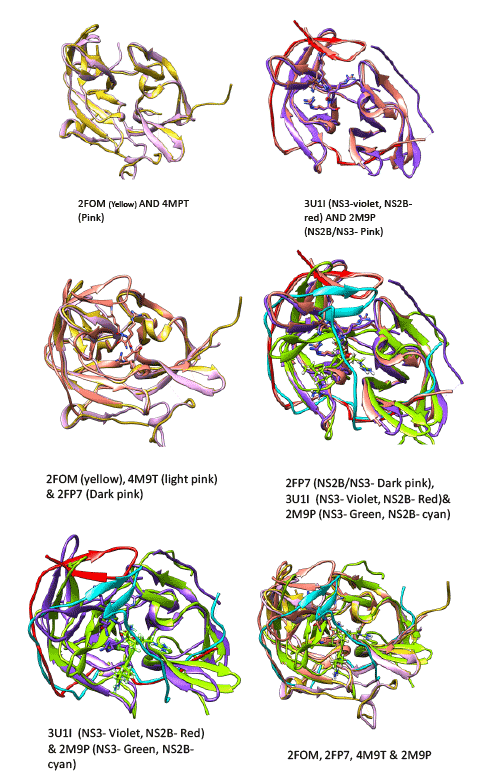
Figure 2: Superimposed structures of 2FOM, 4M9T, 2M9P, 2FP7, 3U1I
The proteins were downloaded from Protein Data Bank (PDB). The peptides were modeled using PyMOL. The three dimensional structures of the natural products from papaya leaves were downloaded from Public chemical database (Pubchem). The three dimensional structures of synthetic compounds were obtained from crystallographic studies. The structure of the ligands has to be optimized in order to rectify steric clashes and also to calculate the minimum potential energy. So, prior to docking, all the ligands were minimized using OPLS 2005 force-field (Optimized Potentials for Liquid Simulations) in the Impact module of Schrödinger 2009. During minimization, the ligands were subjected first to steepest descent for 1000 iterative cycles available in the Impact minimization module. This is a primary step, where the initial ligand geometry with steric clashes effect can be treated properly. This was then followed by conjugate gradient for 5000 cycles which makes good convergence in the structure with respect to energy and gradient. The output of this was chosen for the docking [30]. The protein was minimized using protein preparation wizard where addition of H atoms and bond order were adjusted and further energy minimization was carried out using OPLS2005 force-field. Molecular docking helps in identifying energetically and geometrically favorable binding pose of a ligand bound to the protein. Out of different types of docking, Induced fit docking possess more advantage. This method of docking helps to treat both the ligand and the protein as flexible. Induced fit (Glide XP) module which possesses flexible docking option was used for docking of ligands with protein. The grid was specified for the site of docking by specifying the active site residues, in this case, the catalytic triad residues. Grid of 20 Å along each edge is specified for the calculations to be performed. The resulting output file was analyzed and the best pose was considered for molecular simulation studies. The molecular dynamic simulations were carried out using AMBER 12 (Assisted Model Building with Energy Refinement) [31]. The molecular dynamic simulation helps to analyze the interactions and behavior of the ligand in dynamic state over a time scale. This also helps us to know the stability of the protein-ligand complex. AMBER FF99SB force field was used for the parameterization of the protein molecule. TIP3P water box was used for the solvation of the complex and charge neutralization was carried out using Na+ and Cl- ions. The total complex with water molecules was minimized first and then equilibration was carried out until the system reaches a stable temperature and pressure.
Extracts from different plant sources are known to have medicinal importance. There are many successful cases where compounds and secondary metabolites from the plants have been proved to have antimicrobial activity. In the recent outbreak of dengue in India, there were reports that many medical practitioners have used papaya leaf extract to cure dengue fever and cases of successful treatment were also seen. Extracts from the papaya leaf have been reported to have an antidengue activity [32]. In the light of this we have analyzed the papaya leaf extract using GCMS (Gas Chromatography Mass Spectrometry) and found three secondary metabolites, oleic, stearic and palmatic acids as major constituents. We have then carried out molecular modeling studies of the three compounds towards the NS2B/NS3 protease, the interactions and the glide score were relatively higher and the interaction with active site residues were favorable when the docking was carried out with 2M9P as target compared with 2FOM as the target. The docking score, glide energy and interaction diagrams are presented. All the three compounds possess interactions with the catalytic residues, with one H-bond interaction and some non-bonded interactions also. The region of the binding at the catalytic site for oleic and palmatic acid is similar but it is different in the case of the stearic acid. The information regarding the score, energy and interaction are shown in table1 and the binding sites are shown in the figures 3a and 3b. The ligands oleic acid and palmatic acid were bound in a similar binding mode as in the co-crystal structure, where as the stearic acid has a different binding. The binding orientation of the two ligands which were similar to the co-crystal is represented in figure 3c. The ligand is shown in stick and the protein residues are shown in line format, with the active site residues highlighted in cyan colour. The compounds showed an improved score and energy in the binding with the protein in the modeled NMR structure, where the full structure is modeled with NS2B fragments.
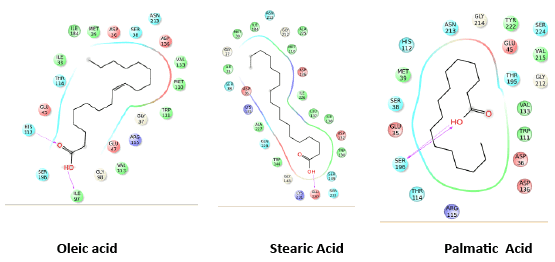
Figure 3a: Interaction of oleic acid, stearic acid and palmatic acid
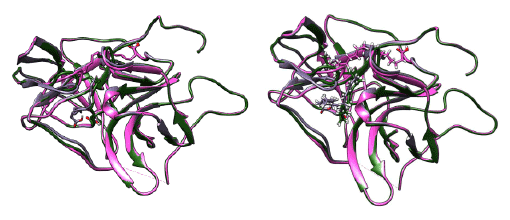
Figure 3b: Binding mode of oleic acid (Green), stearic acid (Pink) and palmatic acid (Purple)
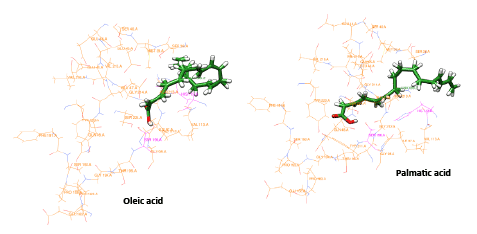
Figure 3c: Binding orientation of Oleic acid and Palmatic acid
The compounds which were synthesized by our collaborators and reported [33] were also subjected to modeling using 3U1I as template. These compounds also showed improved binding, and they were also subjected to dynamic simulations. The analysis showed that the compounds were bound and showed a better interaction during the course of the trajectory.
The use of peptides as favorable drugs than the synthetic compounds has gained much interest, as they are less toxic with minimum side effects. There have been many efforts to design peptides based on the active site structure or based on the substrate. Many peptide leads have been reported in the recent times as potent inhibitors against the dengue protease [34]. These peptides are also end modified to incorporate the influence of the functional groups. From our previous modeling studies we have reported peptides SHMG, GHMS isolated from edible fishes, and designed peptides which can bind with good energy and score [35]. Peptides which showed good results have now been subjected to docking with the 2M9P as target to see if the interaction has been subjected to any change. The binding of the peptides to the target was found to be better with improved score and energy values. The docking score and energy values are tabulated (Table 2) with the interactions shown in figure 4. The binding site of the majority of the peptides is similar to that of the co-crystal ligand in the solution structure. With an effect on the energy but with increase in docking score, the binding of other peptides is not only at the active site of NS3 but their interactions can also be seen with residues of NS2B. These possess hydrogen mediated interactions with SER and HIS and maintain non-bonded interactions with the other active site residues. The presence of proline and glycine in the peptide as reported in the previous modeling papers show an affinity with additional interactions. The orientation of the reverse peptides makes these to interact with both the chains. In view of the peculiar nature of its sequence and its availability in nature (these peptides are isolated from edible fishes), the reverse peptides were subjected to simulation studies. The target bound peptides were subjected to molecular dynamic simulation for 30 ns and showed a consistent binding throughout the trajectory time (data not shown). The position of the peptides and their superimposed information of the individual peptides with the 2M9P are given in the figures 5,6. It can be observed that the binding pocket of the peptides, GHMS and SMHG are similar to that of the co-crystal. The binding of the ligand with the Histidine and Serine amino acids can be found in both the peptides as seen in the co-crystal. The GHMS has a similar orientation in the pocket as in the case of cocrystal. Figures 6a and 6b show the binding orientations of the peptides and co-crystal ligand in the pocket respectively. The active site residues are represented in the cyan colour, the ligands are represented in stick model with the protein residues been projected as line with three letter indicator with residue name and number (table 3).
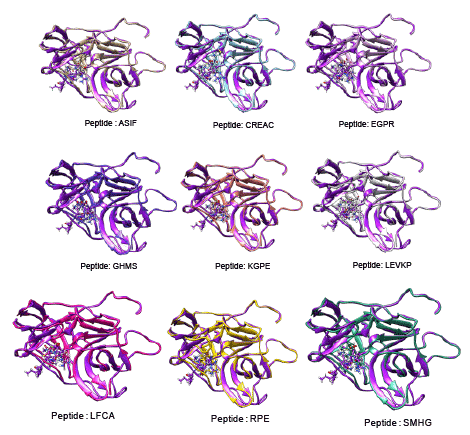
Figure 4: Binding mode of different peptides superimposed with 2M9P (violet)
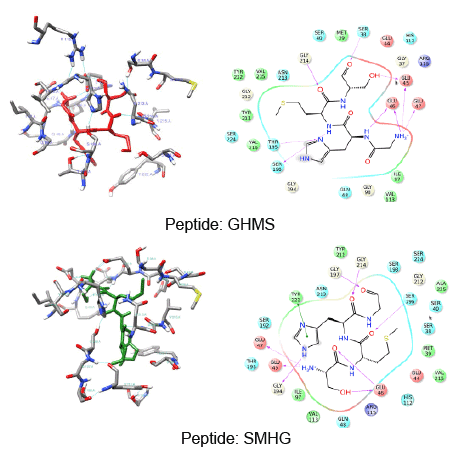
Figure 5: Interactions of Reverse peptides (SMHG and GHMS)
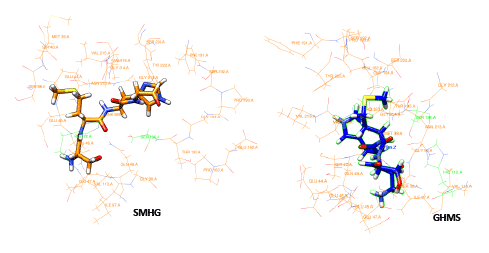
Figure 6a: Binding orientation of SMHG and GHMS
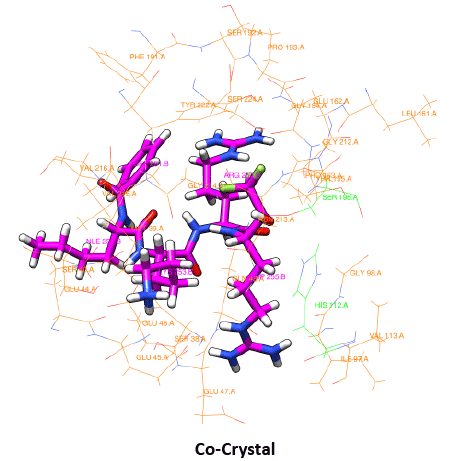
Figure 6b: Binding orientation of Co-crystal
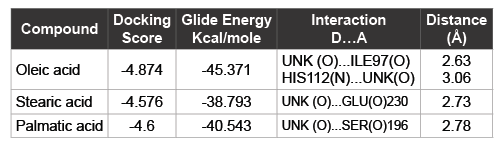
Table 1: Induced fit docking results of compounds from papaya leaf
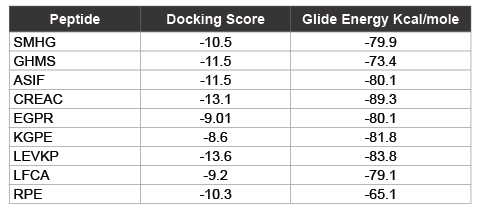
Table 2: Induced fit docking results of Peptides
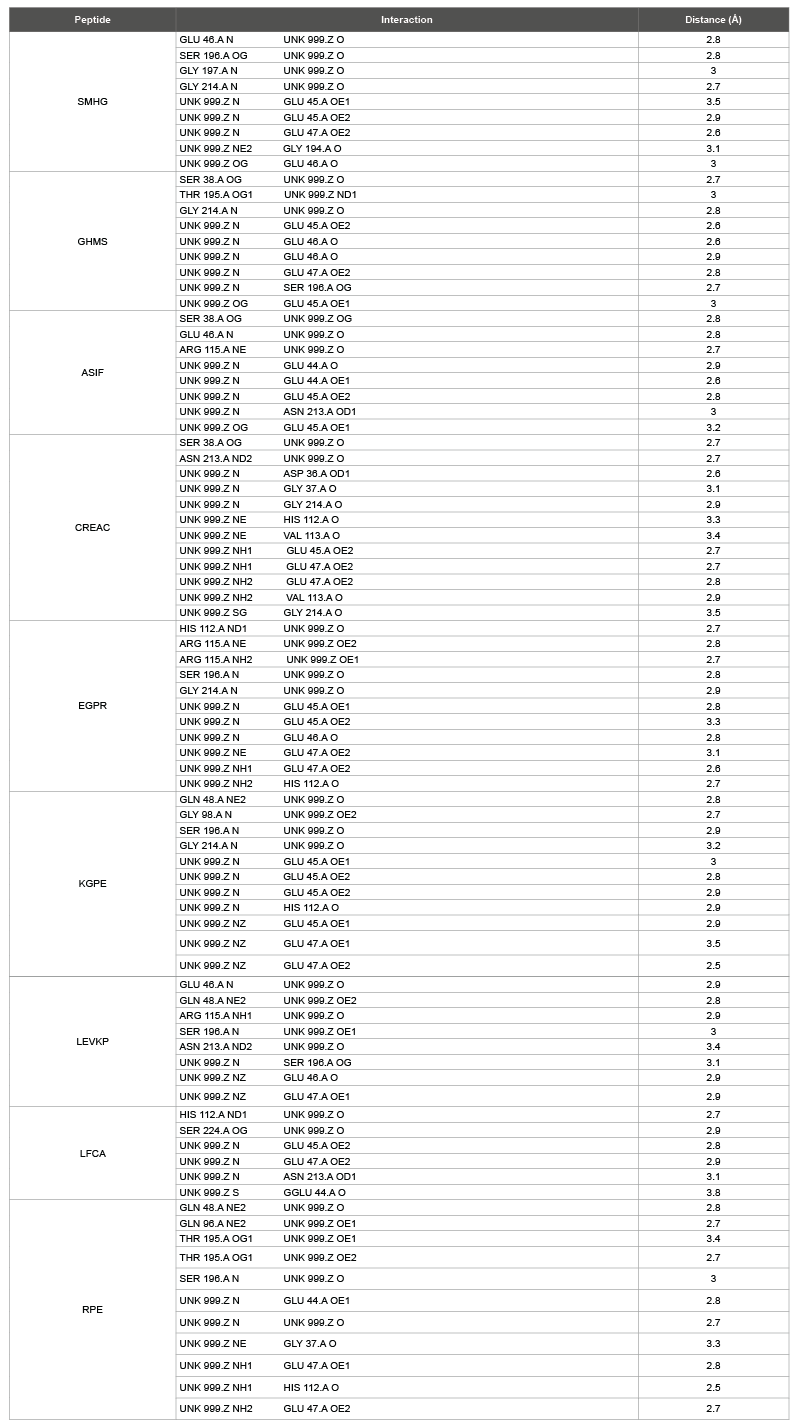
Table 3: Interaction details of the peptides with protease
Our attempt to design compounds against dengue virus protease has not been a straight forward task. Even though the main architecture of the dengue protease is similar to serine protease, the presence of the active site on the surface of the protein, hydrophobic pocket and the influence of the NS2B cofactor have a large influence on the ligand binding. We have designed a series of peptides specific to the protease, through substrate based and active site based approach. Their binding modes towards the protease were analyzed through modeling studies and are validated by subjecting the complex to molecular dynamic simulations. This approach may help to find out not only a static low energetic structure but also a stable complex. This can help in completing the in silico method for the identification of the best possible ligand as an inhibitor. From this particular study, one can observe that the binding efficiency of ligands is influenced by the protein state and the crucial residues, which at times won’t show up in the crystallographic studies. The modeling of the missing residues with help of conserved and similar counterpart will help in choosing the better ligand. Docking and dynamic studies carried out with the structure reported from NMR studies also confirmed improvement in the binding affinity and also the mode of binding. The molecular dynamics further helped us in finding the dynamic nature of the ligand also and its energetics in binding with the active site residues. Encouraged by the above results attempts are underway for cocrystallization of NS2B/NS3 with peptides and also with Oleic acid and Palmatic acid.
The authors wish to thank UGC and DBT (INDO-GERMAN) for the financial assistance.
Download Provisional PDF Here
Article Type: Research Article
Citation: Kutumbarao NHV, Velmurugan D (2016) Structural Analysis and Molecular Modeling Studies of Fatty Acids and Peptides Binding with NS2B/ NS3 Dengue Protease. J Emerg Dis Virol 2(4): doi http://dx.doi.org/10.16966/2473-1846.121
Copyright: © 2016 Velmurugan D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access