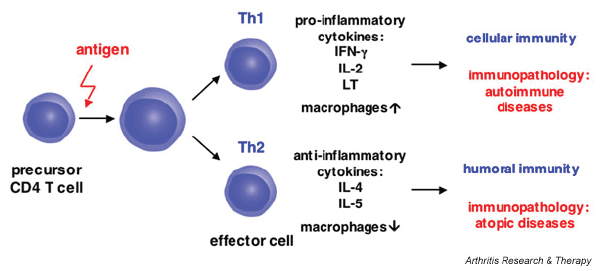
Figure 1: CD4+ differentiation into both Th1 and Th2 cells and their functions
Sefinew Molla*
Biomedical Science Stream, Department of Biology, College of Science, Debre Tabor University, Ethiopia*Corresponding author: Biomedical Science Stream, Department of Biology, College of Science, Debre Tabor University, Deber Tabor, Ethiopia, E-mail: sefinewm21@gmail.com
Hepatitis is a general term for an inflammation of the liver due to a variety of causes including metabolic diseases, drugs, alcohol, toxins, and viruses. Viruses are the most common causative agents of hepatitis today and infect many millions of individuals annually. Viral hepatitis encompasses several diseases and represents a global health problem. It induces major morbidity and mortality and places enormous demands on economic and medical resources. Several viruses that cause hepatitis have been isolated in the past decades: hepatitis A (formerly ‘infectious’ hepatitis), hepatitis B (formerly ‘serum’ hepatitis), and hepatitis C (formerly ‘non-A, non-B’ hepatitis), hepatitis D (delta hepatitis), and hepatitis E (formerly ‘enterically transmitted non-A, non-B’ hepatitis) [1]. Recently, a new virus (hepatitis G or GBV-C) has been identified as a sixth viral agent [2].
Hepatitis B is a small partially double-stranded circular DNA virus that favors replication in liver cells. Therefore, it is termed hepatotropic virus classified in the hepadnaviridae family. Hepatitis B virus is the major cause of liver disease that varies greatly in severity from person to person [3].
HBV causes liver diseases that vary greatly in severity from person to person. Some subjects control infection efficiently and clear the virus from the bloodstream either without clinically evident liver disease or with an acute inflammation of the liver (acute hepatitis) that can resolve without long-term clinical sequelae. Other patients fail to clear the virus and develop chronic infection [4]. Most chronically infected patients remain largely asymptomatic without life-threatening liver disease but 10–30% develops liver cirrhosis with possible progression to liver cancer [5,6]. The rate of HBV chronicity is low in adult infections (5% or lower) but age and route of infection influence the outcome with exposure in neonatal life leading to a high rate of HBV persistence [6,4].
Hepatitis B virus DNA virus characterized by its very small genome size and its unique replication via reverse transcription. The circular genome has been efficiently exploited, thereby limiting genome variation, and leaves no space for genes in addition to those essentially needed during the viral live cycle. Hepatitis B virus is a prototype non-cytopathic virus causing persistent infection. Hepatitis B virus (HBV) infects over 300 million people worldwide and despite the availability of a vaccine, it remains the second biggest carcinogen in the world and a serious global health problem. Human hepatitis B virus (HBV), as well as the closely related animal viruses, most frequently is transmitted vertically from mothers to their offspring. As a persistent virus, hepatitis B virus (HBV) is believed to be non cytopathic in most circumstances, with its disease pathogenesis mediated by host innate and adaptive immune responses. Although HBV may initially avoid activating critical innate intracellular defenses (eg, type I interferon), T cells exert both cytopathic and non cytopathic antiviral effects toward resolution of HBV infection. Because infection usually persists for many years, if not lifelong, hepatitis B viruses need efficient mechanisms to hide from the immune response of the host. To escape the immune response, they exploit different strategies. Firstly, they use their structural and non-structural proteins in multiple ways. One of these is to alter the immune response. Secondly, they replicate and establish a pool of stable extra chromosomal transcription templates, which allow the virus to react sensitively to changes in its microenvironment by up- or down regulating gene expression. Thirdly, hepatitis B viruses replicate in the liver which is an immune privileged site.
HBV is not directly hepatotoxic but its interaction with the host immune system creates opportunity for HBV DNA integration into the host genome. HBV causes liver injury by an immune response against the virus-infected liver cells, although immune suppression appears to enhance replication and induced cytotoxicity. The interplay of the host immune response and the viral ability to replicate is a prime determinant of the likelihood of liver injury, its intensity, and progression to cirrhosis. A series of stages evolve in the life cycle of each patient’s infection, with associated decreases in viral load at each successive stage. Viral mutations in the polymerase or the core gene affect replication and may enhance liver injury. Recently, genotypes have been identified that are linked to clinical outcomes, drug responses, and mutations. Following HBV infection, there is an initial hepatitis that may or may not be symptomatic. Successful clearance and resolution of infection depends on the age and immune status of the individual with most infections of immune competent adults being self-limiting. Persistent or chronic infection is more likely to occur following vertical transmission (from mother to child) or horizontal transmission to children or to immune compromised adults [4]. The immune determinants of successful clearance of HBV are not fully understood but both cellular and humoral immune responses are important [7]. At the same time, however, liver inflammation and disease are also believed to be largely immune-mediated. Therefore, a complex interaction exists between HBV and the host in the initial clearance of HBV, the long-term persistence of HBV and the pathogenesis of HBV liver disease.
HBV has been found in virtually all body secretions and excretions. However, only blood, body fluids containing visible blood, semen and vaginal secretions represent a risk of transmission [8]. Three major routes spread HBV: perinatal, horizontal, and sexual transmission [9]. In developing countries, the main routes of transmission are: neonatal with HBV carrier mother infecting her infant usually during birth or soon after birth following close contact, transfer of HBV via cuts, sexual transmission, transfusion of infected blood or blood products, needle stick injury, contamination of eye, re-use of HBV contaminated needles, syringes, lancets and instruments including those used in tribal ceremonies, possibly blood sucking insects and bed bugs [10].
HBV is transmitted by percutaneous and mucosal exposure to infective blood or body fluids. Major modes of HBV transmission include sexual or close household contact with an infected person, perinatal mother to infant transmission, intravenous drug use and nosocomial exposure [11]. Percutaneous exposures that have resulted in HBV transmission include transfusion of unscreened blood or blood products, sharing unsterilized injection needles for IV drug use, haemodialysis, acupuncture, tattooing and injuries from contaminated sharp instruments sustained by hospital personnel [12].
The global prevalence of chronic HBV infection varies widely, from high (≥ 8%, e.g., Africa, Asia, and the western Pacific) to intermediate (2-7%) e.g. Southern and Eastern Europe) and low (<2%, e.g. Western Europe, North America and Australia [13]. Most countries in Africa have high HBV endemicity, with the exceptions of Tunisia and Morocco, which have intermediate endemicity [14].
HBV infection and its sequelae (cirrhosis and liver cancer) are major global health problems. It has been estimated that up to 2 billion individuals have evidence of exposure to HBV [15] and an estimated 350 million persons worldwide are chronically infected with HBV [6,14]. Most of these come from East Asia and sub-Saharan Africa [16,12]. Approximately 470 million inhabitants of Africa are infected with this virus at some time during their lives and about 10% remain infected. HBV induced diseases, especially hepatocellular carcinoma (HCC), causes more than 230, 000 deaths in African each year [17]. Overall prevalence in Ethiopia varies from 4.7-16.8% for HBsAg and 70-76.38% for at least one positive marker [18-22].
This review will assess interaction of hepatitis B virus with human liver cell and the role of immune cells in the clearance of the virus and its persistence. It also explores innate and adaptive immunity and their components and their role in HBV infection. Finally, it will briefly describe how the virus escapes from immune cells.
Hepatitis B virus (HBV) infection is one of the main causes of chronic liver diseases. The course of viral infection is a balance between the virus and the host immune defense. The host-virus interactions are further complicated by viral immune evasion mechanisms and host immune regulatory mechanisms. It is widely accepted that adaptive immune responses play major roles in the defense of HBV infection [23]. However, the role of innate immunity during HBV infection appears not to be well understood [24]. Increased knowledge of innate immunity will allow us to illustrate the role of innate immune cells and pattern-recognition receptors (PRRs) involved in viral clearance and persistence.
Our primary innate immune cells include natural killer (NK) cells, natural killer T (NKT) cells, and macrophages [24]. Sometimes dendritic cells, esp. plasmacytoid DCs (pDCs), are also included [25]. They consist of the first defense line against invading pathogens and sense danger signals. PRRs are widely expressed on innate immune cells, and are grouped into Toll-like receptors (TLRs), nucleotide-binding oligomerization domain leucine-rich repeat proteins (NODs), and RIG-Ilike receptors (RLRs) [26,27]. Once the PRR senses pathogen-associated molecular patterns from microbes, the intracellular signals are triggered and pass finally leads to production of interferon (IFN)-α/β, IFN-γ and inflammatory cytokines [28].
The liver is an immunological organ with a predominant innate immune system. Innate immune cells and PRR not only sense HBV infection and respond immediately to clear virus at the initial period of infection, but also help to initiate adaptive immune responses to viral infections [29]. To escape the surveillance of the immune response, HBV develops strategies to suppress the antiviral immune response by generating viral partners to inhibit the induction of antiviral genes. Also, HBV-induced immune tolerance is thought to be the main cause of unsuccessful clearance of HBV [30,31]. Although activation of innate immune responses exerts key roles in potential inhibition of HBV replication [29,32], over-activation of immune cells or PRRs signal pathway may lead to liver injury.
The outcome and progression of HBV infection are determined by interactions between virus virions and host immune system. Achieving successful clearance of HBV requires effective viral suppression, the activation of innate immunity, the evoking of antigen-specific adaptive antiviral immune response, and the reverse of immune tolerance [24]. Therefore, comprehensively understanding the interaction between HBV and the host immune system, particularly the mechanisms of HBV recognition and clearance by the immune system and the suppression of HBV to immune responses are helpful in the design of effective therapeutic strategies for chronic HBV (CHB) infection.
An efficient control of viral infection requires the coordinated action of both innate and adaptive immune responses. Innate immunity has evolved to rapidly recognize viral nucleic acids, viral proteins and tissue damage. It induces an antiviral state on infected cells by producing type I interferon (IFN), decreases the pool of infected cells by directing natural killer (NK) cell-mediated killing of viral infected cells, and supports the efficient maturation and site recruitment of adaptive immunity through production of pro-inflammatory cytokines and chemokines [33]. These mechanisms reduce virus spread until the adaptive branch of immunity takes the stage.
As a foreign pathogen enters the host, it is immediately detected by pathogen-sensing mechanisms such as the pattern recognition receptors (PRR) through its pathogen associated molecular patterns (PAMPs), thereby activating the innate defense system [34,35]. Innate immunity generally plays a role immediately after infection to limit the spread of the pathogen and initiate efficient development of an adaptive immune response. Innate host responses during the early phases of viral infections are mainly characterized by the production of type 1 interferon (IFN)-α/β cytokines and the activation of natural killer (NK) cells. Production of type 1 IFNs can be triggered directly by virus replication through cellular mechanisms that detect the presence of viral RNA or DNA [36-38], while NK cells are activated by the recognition of stress-induced molecules and/ or the modulation of the quantity of major histocompatibility complex (MHC)-class I molecules on the surface of infected cells [39].
Following infection with HBV, it is believed that hepatocytes, which have low expression of human leukocyte antigen (HLA) class I, release IFN-α and IFN-β. Initial recognition of HBV infection may be mediated by toll-like receptors (TLRs) [40]. In HBV transgenic mice, the production of IFN-α/β is associated with a 10-fold reduction of viral capsids containing HBV pregenomic RNA and the activation of double-stranded dependent protein kinase activity (PKR), which inhibits HBV protein synthesis [41]. In addition to this, IFN-α/β recruits and mediates the activities of antigen presenting cells (APCs), in particular Kupffer cells (macrophages that reside in the liver) and dendritic cells (DCs). These APCs in turn produce interleukin-18 (IL- 18) and the chemokine CCL3, which induces natural killer (NK) and natural killer T (NKT) cell activity [42]. NK cells, NKT cells and Kupffer cells have all been shown to play a role in the initial response against HBV [43].
The initial phase of innate immune responses in human HBV infection is not well-characterized because of practical difficulties in identifying patients immediately upon HBV exposure and inaccessibility of relevant tissue compartments (eg, site of virus entry, lymph nodes, liver) [44]. The activity of NK and NKT cells is likely to be an important anti-HBV response that precedes the up regulation of HLA class I expression on hepatocytes. Up regulation of HLA class I expression is critical for presentation and recognition of foreign antigens by T-cells in the adaptive arm of the immune response [45]. Therefore, the innate immune system likely controls HBV replication in the early stages of infection before the detection of any hepatic inflammatory cell infiltrates or associated liver damage.
Despite the lack of innate type I IFN response, HBV infection is resolved in most adults. However, HBV is readily controlled by IFN-α [46] as well as TLR, NK, NKT, and APC [47]. HBV can activate innate immune cells such as Kupffer cells [48] and NK/NKT cells. For example, dynamic changes in NK and NKT cell activation were observed in careful monitoring of two patients identified early during acute HBV infection [49], whereas another study showed an early production of regulatory cytokine IL-10 associated with attenuated NK and T-cell response [50]. Thus, HBV may avoid the type I IFN response but modulate other innate immune pathways during acute infection.
HBV persistence is associated with changes in innate immune parameters. For example, NK cells from patients with chronic hepatitis B display reduced cytotoxicity and cytokine production upon stimulation [51]. Moreover, increased serum IFN-α level was observed during hepatitis flares in patients with chronic hepatitis B [52], unlike in acute hepatitis B. The underlying mechanism for differential type I IFN response between acute and chronic HBV infection is not clear. However, elevated serum IL-8 levels (as well as IFN-α) during chronic hepatitis flares may contribute to liver injury by up regulating NK expression of TNF-related apoptosisinducing ligand (TRAIL) and hepatocyte expression of TRAIL deathinducing receptor. Chronic hepatitis B is also associated with mDC and/or pDC dysfunction that may be reversed by therapeutic virus suppression [53]. Collectively, multiple innate immune pathways are induced during acute and chronic HBV infection, thus shaping the course of infection.
Adaptive immune response is mediated by B and T cells. The adaptive immune response is induced after the initial innate immune response and it targets specific antigenic sequences (epitopes) encoded by the pathogen. It can also be maintained after primary infection, providing pathogenspecific protective immunity against reinfection.
B cells produce antibodies and mediate the humoral adaptive immunity and cell mediated immunity which involves the association of T-cells with antigen presenting cells are the main components of adaptive immune system. Antibodies consist of an antigen binding variable (V) region and a constant (C) region with the Fc region, which can bind cellular receptors and complements [54]. Neutralizing antibodies can limit viral spread by directly binding and removing the circulating virions or by blocking target cell entry. However, antibodies can also contribute to inflammatory response, enhancing antigen presentation and binding pro-inflammatory microbial products. Regulatory B cells (Bregs) secreting immune regulatory cytokine IL-10 have been described [55].
Helper T-cells (CD4 T cells) recognize cells presenting exogenous peptide antigens in the context of class II MHC molecules. Class II MHC is limited to certain specialized immune cells (eg, dendritic cells, B cells, and macrophages), but not hepatocytes. Therefore, CD4 T cells may not be readily activated by hepatocytes [56]. However, CD4 T cells play an important immune regulatory role, activating dendritic cells to prime CD8 T cells, producing cytokines, and providing T-cell help for B cells. By contrast, CD8 T cells recognize cells expressing endogenously synthesized peptide antigens in the context of class I MHC. Because most cells express class I MHC (including hepatocytes), CD8 T cells can directly recognize and kill virus-infected cells while exerting noncytolytic antiviral effect by cytokines [47].
Humoral immune responses against HBV infection: Antibody response to HBV envelope (anti-HBs) has virus neutralizing capacity and is associated with HBV clearance and protective immunity [57]. Accordingly, passive administration of hepatitis B immunoglobulin provides post exposure prophylaxis and prevents graft infection in HBV infected liver transplant recipients. However, anti-HBs are detectable relatively late as acute hepatitis resolves and the circulating HBsAg level declines [47]. This raises the possibility that anti-HBs is not critical for HBV clearance in primary infection. Alternatively, anti-HBs may be induced and mediate HBV clearance. However, it may not be serologically detectable because it is complexed with circulating HBsAg and cannot be detected serologically until the HBsAg level drops.
Antibody responses to each of the HBV proteins have been detected in the sera of humans following transient HBV infection. Anti-HBs antibodies are a marker of resolution of transient HBV infection. It is generally accepted that neutralizing anti-HBs antibodies plays a key role in recovery from infection with HBV by containing the spread of infection in the infected host and facilitating the removal and destruction of viral particles. These antibodies have also been shown to prevent reinfection by blocking the ability of virus particles binding to receptors on target cells. In chronic HBV infection, antibodies to the viral surface proteins are generally not detected in serum although it is possible their presence is masked by the formation of immune complexes with surface antigen particles present in the bloodstream [58].
The humoral response is also critical to long-term clearance of HBV and protection from infection with HBV. In patients who recover from acute HBV infection, activated T-helper cell type 2 (Th2) CD4+ T-cells induce B-cell production of HBsAb, HBcAb and HBeAb. HBsAb are synthesized early in infection but are not detectable because they are complexed with the excess of envelope antigens produced during virus replication [59].
Cell mediated immune responses against HBV infection: One of the challenges in understanding viral pathogenesis is the elucidation of the full repertoire of immune responses that control the replication of the invading pathogen. Generally speaking, such control mechanisms can be either noncytocidal or cytocidal. Noncytocidal responses can result from the release of cytokines or other substances with antiviral potential, without necessarily injuring the infected cell. By contrast, cytocidal responses aid in suppressing the pathogen but can also contribute to tissue injury and disease via killing infected cells.
The T-cell response during acute self-limited hepatitis B in people is characterized by a vigorous, polyclonal, and multi specific cytotoxic and helper-T-cell response. Although clearance of most virus infections is widely thought to indicate the killing of infected cells by virus-specific T cells, it was suggested that non-cytolytic intracellular viral inactivation by cytokines released by virus-inactivated lymphomononuclear cells have an important role in the clearance of this virus without killing the infected cells [60]. Recent studies using a transgenic mouse model of hepatitis B virus infection have also shown that adoptive transferred, virus-specific cytotoxic T cells can abolish hepatitis B virus gene expression and replication in the liver without killing the hepatocytes [61]. Additional factors that may contribute to viral persistence are immunological tolerance to viral antigens, viral inhibition of antigen processing or presentation, infection of immunologically privileged sites, modulation of the response to cytotoxic mediators, and viral mutations [62].
The three structural forms of the viral proteins, the HBsAg, the particulate HBcAg, and the non particulate HBeAg, may preferentially elicit different Th cell subsets. In previous studies, HBeAg has been shown to induce a Th2 immune response in mice, whereas HBcAg induced a Th1 response [63]. The Th2 response to the HBeAg was dominant over the Th1 response to the HBcAg, resulting in the depletion of HBcAg-specific Th1 cells in vivo [64]. It is also accepted that cell mediated immune responses are important in the elimination of viruses that do not have a lytic cycle in the host and for any tissue damage seen during either transient or persistent infection, or both. In the direct elimination performed by killer T cells, the cell mediated immune response also needs assistance from the cells, which work in two ways. First, the Th1 cells stimulate macrophages, which then clear virus particles. Second, the Th2 cells stimulate B cells to generate immunoglobulins, which adhere to the surface of virus particles and induce opsonization. It has been shown that low doses of virus were able to induce a protective cytotoxic T lymphocyte (CTL) response (Th1- mediated), whereas high doses of virus failed to do so and induced a nonprotective humoral (Th2-mediated) response instead [65].
Regulation of HBV occurs through non-cytolytic down regulation of viral replication, which means that viral clearance results without killing the infected cells. Generally, this occurs through the release of cytokines by virus-inactivated lymphomononuclear cells [66]. As a result, virusspecific CD8+ and CD4+ T cells play key effector and regulatory roles in hepatitis B antiviral immunity. Though CD8+ T cells are the main effector cells that cause viral clearance, CD4+ T cells are necessary to facilitate the induction and maintenance of CD8+ T cells [47].
CD4+ T cell response: Activated HBV-specific CD4+ T cells are multi specific. However, strong helper T cell responses are found against certain peptides in both the HBcAg and HBeAg following resolution of acute HBV infection. While HBeAg has been shown to induce a Th2 immune response, HBcAg stimulated a Th1 response. Additionally, CD4+ T cell responses are detected against polymerase and X antigens [67]. Because the Th2 response to HBeAg was dominant to the Th1 response to the HBcAg, the HBcAg-specific T cells were depleted in vivo [3].
It is important that both Th1 and Th2 responses are generated because they induce different responses. While the Th1 cells stimulate macrophages, which can clear virus particles, the Th2 cells stimulate humoral immunity, in which B cells are stimulated to generate immunoglobulin for opsonization. However, different doses of virus seem to generate different responses. For example, low doses of virus produce a Th1-mediated response while high doses of virus produce a Th2-mediated response [3]. The figure below shows a flow chart of CD4+ differentiation into both Th1 and Th2 cells and their functions (Figure 1).
Figure 1: CD4+ differentiation into both Th1 and Th2 cells and their functions
CD8+ T cell response: CD8+ T cells are the main effector cells in the viral clearance of HBV. Experiments that depleted CD8+ T cells in chimpanzees after infection with HBV resulted in viral persistence of HBV, indicating the importance of these cells. CD8+ T cells produce IFN-γ, which clear HBV through destabilization of the viral capsid, degradation of viral proteins, and post-transcriptional degradation of HBV RNA. The combined effect of cytokines, such as IFN- γ (which can also be produced by HBV specific Th1 CD4+ T cells), and cytolytic activity leads to the destruction of the virus without excessive liver damage [67]. Figure below shows a CD8+ cell interacting with an infected cell (Figure 2).
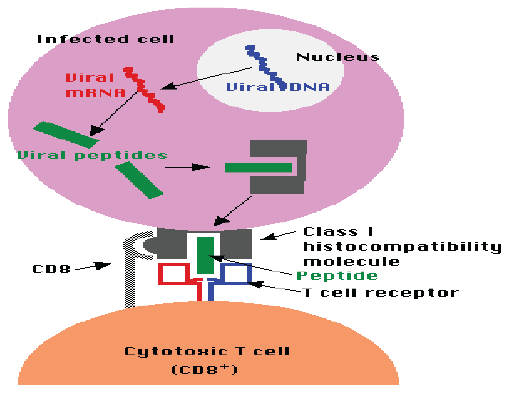
Figure 2: CD8+ cell interacting with an infected cell
“Professional” antigen presentation cells comprise the MHC-class II-expressing B cells, macrophages and dendritic cells (DC). It can be hypothesized that the deficient T cell responses associated with HBV infection may be linked with deficiencies in the APC and impaired T cell priming.
DC are the most important APC and these cells take up and process antigen and then, migrate to the lymphoid organs where they present the antigen to the T cell [68]. DC is generated from precursor cells e.g. monocytes or macrophages, via signals involving cytokines such as IL-4 and granulocyte-macrophage-colony stimulating factor (GMCSF). Upon their generation, DC are said to be immature and serve a phagocytic function until they receive the correct signal which stimulates their differentiation into a mature phenotype. For instance, in viral infection, the recognition of viral double stranded RNA can trigger the differentiation of DC. Once mature, DC upregulate numerous adhesion and antigen presentation molecules and then migrate to the lymph node where their primary function is antigen presentation. They also produce numerous cytokines to recruit other arms of the immune response [69].
The findings concerning dendritic cells (DC) in HBV are conflicting. Defective function and immature phenotype of DC has been observed in HBV infection [70,71]. However, Tavkioli et al. [72] studied monocyte derived DC (MDDC) and found that while there were a few minor phenotypical alterations and slightly reduced IL-12 production by MDDC in HBV, their T cell stimulatory capacity was unaffected. Another study found that the expression of co-stimulatory molecules on DC and their capacity to stimulate T cells was impaired in chronic HBV infection (CHB) but could be restored using a cytokine cocktail [71]. In 2007, Tavakioli et al. [72] published their research on myeloid DC and plasmacytoid DC in which they found no quantitative, phenotypic or functional defects in chronic HBV carriers, compared to uninfected control subjects. The role of DC has not yet been elucidated in the clearance or persistence of HBV infection, however, IFN-α treatment has been shown to increase frequencies of circulating DC and increase the expression of HLA-DR, CD80 and CD54 (ICAM-1) by such cells [73]. One interesting report found that plasmacytoid DC from patients chronically infected with HBV induced the generation of a higher proportion of CD4+CD25+ Treg cells compared to those from uninfected controls or HBV resolvers [74].
Macrophages are a second type of “professional” APC. They are longlived, phagocytic cells that circulate in blood as monocytes & reside in organs & tissues. These phagocytic cells engulf microorganisms, red cells, immune complexes and endotoxins and present the processed antigens to T cells [75]. Once activated by infected viruses or by NK, NKT or T cell derived cytokines, macrophages can produce cytokines that have direct (IFN-α/β, TNF-α, nitric oxide) or indirect (IL-1, 6, 8, 10, 12, 08, GM-CSF ) antiviral effects [75,76]. Macrophage-derived IFN-α/β, TNF-α, IL-12 and nitric oxide can inhibit HBV gene expression and replication in HBV transgenic mice [77].
B cells are the third type of “professional” APC. They internalize specific antigen byreceptor-mediated endocytosis and subsequently present the antigen to helper T cells which can then stimulate antibody production by the B cells. The central role of B cells is in the antibody response which is discussed below. B-cell mediated immune responses are crucial to the elimination of HBV.
Cytokines play a key role in the defense against viral infections, both directly, through inhibition of viral replication, and indirectly, through determination of the predominant Th1/Th2 pattern of host response. However, in the context of an inflammatory response against a virus, cytokines may also lead to liver damage [78]. The importance of this is best demonstrated in HBV. In acute HBV infection, a vigorous polyclonal cellular immune response is critical; thus Th1 cytokine release is essential to initiating an effective immune response. Among the cytokines, IFN-γ is one of the most important mediators in the immune system. It is also known to exert inhibitory effects on viral replication [79] and has been generally considered to show more strict species specificity than IFN-α. The cytokines released by CD4+ and CD8+ cells also play an important role in down-regulation of HBV replication, demonstrating that it is possible to control a viral infection without the death of infected cells [80]. However, if there is a defect in the acute response, HBV becomes chronic and consequently the presence of an ongoing suboptimal inflammatory response can activate the process of hepatic fibrosis.
HBV may have specific mechanisms to inhibit cytokine production, highlighting the critical role of these molecules in recovery from infection [81]. On the other hand, there are some of the immune evasion strategies adopted by HBV. These include the antagonism of immune function through the use of homologues of cytokine receptors, expression of viral proteins which interact with cytokine signal transduction and expression of cytokine mimics and host proteins that influence the Th1 and/or Th2 cytokine responses. These immune modulatory strategies can protect the host from the lethal inflammatory effects as well as inhibit the local inflammatory response elicited to kill the HBV. Other strategies include the alterations in cytokine expression such as demonstrated with the HBcAg and terminal protein which can inhibit IFN-β gene expression [82].
Knowledge of the molecular virology of the hepatitis viruses and the responses they elicit has emphasized the importance of host immunity in resolving infection and mediating liver damage. Many viruses cause cytolytic infections in which viral replication occur at the expense of host cell viability. However this is a shortsighted strategy for the virus as it provides a clear “danger signal” that alerts the host’s innate and adaptive immune defense to eliminate the virus and terminate the infection. Such a life cycle requires a high rate of transmission from host to host, causes acute tissue damage and is unlikely to result in persistent infection.
In order to cause chronic infection viruses must use strategies that enable them to evade or modify host immune responses sufficiently to prevent clearance. Of the hepatitis viruses only hepatitis B (HBV) and hepatitis C (HCV) viruses cause chronic infections and in order to do so they must evade host immune responses. Neither hepatitis A virus nor hepatitis E virus cause chronic infection and must be assumed to lack the ability to escape immune responses. Understanding the mechanisms used by HBV to evade host immunity is central to understand pathogenicity and necessary for the development of effective therapeutic strategies. Although knowledge of the mechanisms of immune escape by hepatitis viruses is increasing, considerable insight has come from the study of other viruses, some of which can cause hepatitis such as Epstein-Barr virus (EBV), HIV, and model systems such as murine lymphocytic choriomeningitis virus (LCMV), as well as transgenic mouse models of HBV infection. In this seminar paper I am interested to describe mechanisms that HBV used to escape from human immune defense.
Hepatitis B viruses are prototype non-cytopathic viruses causing persistent infection. Human hepatitis B virus (HBV), as well as the closely related animal viruses, most frequently is transmitted vertically from mothers to their offspring. Because infection usually persists for many years, if not lifelong, hepatitis B viruses need efficient mechanisms to hide from the immune response of the host. To escape the immune response, they exploit different strategies. Although the mechanism of HBV persistence is not fully understood, researchers speculate that chronic HBV results from multiple factors, including HBV-immune suppression, persistence of stable forms of HBV, and infection of immunologically privileged sites [67].
Firstly, they use their structural and non-structural proteins multiples. One of the purposes is to alter the immune response. Secondly, they replicate by establishing a pool of stable extra chromosomal transcription templates, which allow the virus to react sensitively to changes in its microenvironment by up- or down regulating gene expression. Thirdly, hepatitis B viruses replicate in the liver which is an immune privileged site.
Viruses may escape immune recognition by sequestration in sites that are inaccessible to the immune system. HBV encodes a reverse transcriptase that enables the virus to integrate its own DNA within the host genome, becoming invisible to the immune system in the process.
Classic examples of immunologically privileged sites are tissues through which lymphocytes traffic in small numbers, if at all, such as the anterior chamber of the eye, the brain and testis. Studies in transgenic mice have shown that the access of T lymphocytes to hepatocytes expressing HBV proteins is limited by the density of the liver parenchyma even though some T cells clearly do make limited contact with HBV infected cells. It has been established that HBV may be found in many tissues throughout the body but micro vascular barriers, which are not present in the liver, may prevent hepatitis B surface antigen (HBsAg) specific cytotoxic T lymphocytes (CTL) from accessing and attacking HBsAg expressing cells in the kidney or pancreas. Although sequestration of virus in these sites does not result in immune mediated organ damage it provides a potential reservoir of virus that may repopulate the liver, facilitating viral persistence in the face of low level clearance from the liver [83].
Another method of evading immune detection is through the loss of antigenicity. Mutations within the pre-core/core genes of HBV can result in loss of expression of the hepatitis e antigen (HBeAg), effectively removing one of the key targets for the immune response and “hiding” the virus [84]. Antibodies directed against viral antigens lead to immune clearance in many infections. In acute HBV infection antibodies that bind HBV envelope proteins, including HBsAg, lead to clearance. This antibody response is a T cell dependent process. Antibodies directed against HBsAg are thought to complex with free virus particles, removing them from the circulation and possibly preventing their attachment and uptake into susceptible cells. They also contribute to many of the extra hepatic syndromes of HBV infection. A number of reports have described the emergence of HBV strains with mutations in the envelope gene that lead to a loss of detectable HBsAg expression and viral persistence in the face of anti-HBsAg antibodies.8-10 Antibodies to HBV nucleocapsid antigens HBeAg and HBcAg are found in both acute and chronic infection and do not seem to neutralize viral infectivity.
The easiest way to understand immune evasion by HBV is to look at individuals with chronic HBV. Individuals presenting with chronic HBV infection demonstrate diminished CD4+ and CD8+ T-cell responsiveness. It is suspected that low T-cell levels result from high levels of HBeAg, a secretory form of the nucleocapsid antigen, whose function in HBV infection is not well understood. Because HBeAg is not required for viral infection, replication, or assembly, but is still found in all hepadnaviruses, researchers believe that it may have a role in viral persistence [66]. During HBV replication HBeAg is produced in excess and is suspected to have a tolerogenic effect. Often, core-specific T cells are almost undetectable and have a decreased ability to produce IFN-γ. Despite their low responsiveness, CD8+ T-cells are found in the liver where they may cause an inflammatory response but fail to clear the virus [67].
CD4+ T-cells also demonstrate low levels of response in individuals with chronic HBV. One possible reason for this hypo responsiveness is an impaired function of dendritic cells. This notion is controversial, however, because the effect of chronic HBV on dendritic cells seems minimal. Another possible cause of this weak HBV-specific T-cell response is the role of regulatory T (Treg) cells, which have demonstrated suppression of immunological responses against self and foreign antigens through specific cytokines and direct cell to cell contact. Studies have shown that depletion of Treg cells in patients with chronic HBV infection have experienced increased function of HBV-specific T-cells. This too is controversial, however, because the same effects were seen in patients with resolved HBV infection.
Other viral factors, such as the HBx protein, may lead to viral persistence of hepatitis B. HBx is known to modify various cellular pathways including NFκB, which may subsequently alter antigen presentation and affect the immune response. Additionally, HBx may up regulate the expression of HLA class 1 molecules on hepatocytes, recruiting T-cells to the liver and causing continued liver damage [67].
Another possible mechanism of HBV persistence includes the infection of peripheral tissues that cannot easily be reached by lymphocytes and is ignored by the immune system. Such sites are termed immunologically privileged sites. In such places, viruses can persist without recognition by the immune system. Virions released from these sites, however, can infect liver cells and stimulate memory T-cells, perpetuating liver disease [59]. Finally, epitope mutations facilitate HBV persistence by anergizing B and T-cells rather than stimulating their proliferation [47]. The figure below shows a section of a liver damaged by chronic HBV.
HBV can trigger various innate and/or adaptive immune responses. CD4 T cells are needed to regulate the overall adaptive immune response, whereas CD8 T cells are the critical effectors that mediate viral clearance and liver injury. Multiple immune evasion mechanisms may contribute to viral persistence and immune tolerance with T-cell dysfunction. Antiviral T cells are functionally impaired in HBV persistence, but may recruit inflammatory cells that can amplify liver injury with progressive fibrosis and hepato carcinogenesis. While therapeutic options evolve for HBV, better understanding of its immune pathogenetic mechanisms will help to develop better approaches to prevent and treat HBV infection and its considerable disease burden. In general, the immune responses that terminate viral infection work in several steps.
During the early phases of acute viral infection, natural killer (NK) cells and natural killer T-(NKT) cells are the first lines of defense, and the activation of these cells helps to reduce the viral load through the secretion cytokines, such as IFN-g. In the liver, the frequencies of NK and NKT cells are higher than in other organs, and these cells mediating innate immunity are thought to play certain roles in the pathogenesis of HBV infection. Indeed, during the early phases of acute HBV infection, the activation of NK cells helps to reduce the viral load through the action of IFN-g. However, this activation is rapidly suppressed by interleukin (IL) at the peak of viremia, indicating that the roles of NK cells on HBV regulation are rather limited. In fact, in chimpanzees infected by HBV, the innate immune cells, such as NK and NKT cells, do not significantly contribute to the pathogenesis of viral hepatitis during the symptomatic phase of acute or chronic HBV infection. Therefore, it is assumed that a crucial role in the pathogenesis of HBV infection and the control of HBV replication is played by the adaptive immune response primed after the initial response by the innate immunity. From the host’s point of view, clearance of the virus will be preferred and the host’s immune system will therefore try to destroy the virus. The virus, in contrast, is optimized to coexist with the host to allow sufficient progeny production to infect the next host. In hepatitis B viruses, a well balanced replication strategy avoids major pathogenic effects and ensures an intimate cross talk between virus and host.
Download Provisional PDF Here
Article Type: Review Article
Citation: Molla S (2016) Review on Human Immune Response against Hepatitis B Virus (HBV) Infection. J Emerg Dis Virol 2(2): doi http://dx.doi.org/10.16966/ jedv.117
Copyright: © 2016 Molla S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access