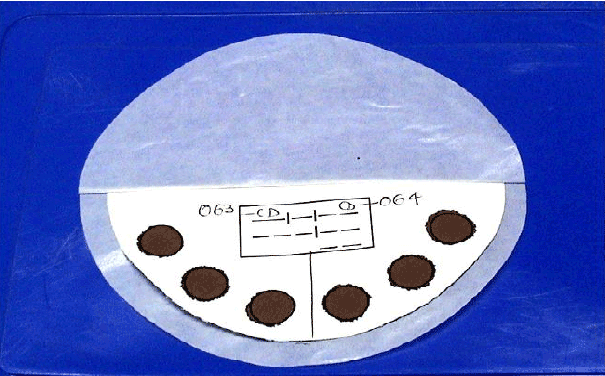
Figure 1: In house developed Filter paper device for collecting Dried Blood Spot.
Lakshmi V*1 Neeraja M1 Lavanya V1 Priyanka EN1 Sharma S2 Dash PK2 Parida MM2
1Department of Microbiology, Nizam’s Institute of Medical Sciences, Panjagutta, Hyderabad, TS, India*Corresponding author: Lakshmi V, Professor & Head, Department of Microbiology, Nizam’s Institute of Medical Sciences, Panjagutta, Hyderabad, 500082, Telangana, India, E-mail: lakshmi57vemu@gmail.com
Detection of Hepatitis C virus (HCV) RNA, the earliest marker and a direct indicator of ongoing viral replication, is more reliable than the anti HCV antibodies for screening for an ongoing HCV infection especially in high risk patients, such as those on hemodialysis (HD). Newer molecular methods that are affordable with a potential for point-of-care testing are being increasingly developed and recommended for an early and cost effective detection of HCV RNA.
An in house developed HCV Reverse Transcriptase Loop mediated Isothermal Amplification (RT-LAMP) assay targeting the 5’ un-translated region (UTR) of the HCV genome, was performed simultaneously on Plasma and Dried blood spots (DBS) from a study group of 300 high-risk patients {250 hemodialysis (HD) and 50 Chronic Liver Disease (CLD)} and 50 healthy age matched individuals (control group). Among the study group, 145/300 (48.3%), (110/250 (44%) HD and 35/50 (70%) CLDs) were positive for HCV RNA on the DBS. The HCV RT-LAMP was 100% sensitive and specific with a % CV less than 10, as compared to the gold standard HCV Real Time PCR and a nested RT-PCR. The detection limit of all the assays was 50 copies of RNA /ml.
Dried Blood Spots (DBS); Reverse Transcriptase; Real time PCR; Hemodialysis
Hepatitis C virus (HCV) infection is an unrecognized hidden epidemic and a major public health problem of global importance [1]. As per the global disease burden, there are an estimated 115 million anti-HCVpositive individuals and 80 million viremic hepatitis C patients [2].
Since an early diagnosis of HCV infection helps to successfully reduce the burden of HCV infection, in the community, an algorithm for a one time mass screening for an early and definitive identification of HCVinfected individuals has been recommended [3,4]. Combined with new and recommended treatments, screening for active infection by detecting HCV RNA would have a great potential to diminish the burden of HCVrelated disease, especially among high-risk individuals [5-7].
The qualitative, non-quantitative target amplification techniques for HCV RNA, such as Reverse-Transcription Polymerase Chain Reaction (RT-PCR) and the Real Time PCR that are now available commercially, are recommended for screening and prevalence purposes due to their lower limits of detection (LOD) of RNA [8].
A nested PCR after the initial RT step to increase assay sensitivity has been developed [9]. However, despite their obtainable magnitude and rapidity of amplification, the RT- PCR assays for HCV are not widely used in remote and resource poor settings as routine diagnostic tools as they require high precision instruments and sophisticated infrastructure [10,11].
Hence, there is a need for development of technically simpler molecular tests that are efficient and rapid as the existing PCR assays, for the detection of HCV RNA, that would be applicable in remote and resource - limited settings, especially in developing countries [10]. Such assays would improve accessibility, affordability and help reduce the burden of HCV in the community [12].
The Loop mediated Isothermal Amplification (LAMP) assay is an efficient, rapid and technically simpler molecular assay [13]. This novel technique for DNA amplification relies on strand-displacing DNA synthesis performed using the Bst DNA polymerase under isothermal conditions (62-65°C) in a simple water bath. The method can be made applicable to RNA genomes by including an initial reverse transcription step [14].
In this study, an RT-LAMP assay targeting the 5’ UTR of the HCV genome was developed and tested simultaneously on plasma and DBS from 300 high risk patients. The performance characteristics of the assay were compared with a nested-HCV RT PCR and a HCV Real Time PCR, available commercially.
The Institutional Ethics Committee of Nizam’s Institute of Medical Sciences, Hyderabad, India (EC/NIMS/957/2008), approved the study. Written informed consent was obtained from each individual included in the study.
Over a period of five years (2009-2013), 300 high-risk patients (250 patients on HD and 50 patients diagnosed to have CLD) and a control group of 50 healthy age matched individuals were included in the study.
Whole blood: Whole blood (3 ml in an EDTA Vacutainer tube, Becton & Dickenson, USA) was collected from both the groups. The plasma were separated from the respective tubes and stored in sterile vials at -20°C until further testing.
Plasma and DBS samples from patients with known Hepatitis B viral infection, Hepatitis A viral infection and Chikungunya infection were also collected.
Dried blood spots (DBS): DBS were collected from all subjects of the 2 groups through a finger prick. From each patient or control, 6 DBS (each 50 µl) were collected on six 12 mm pre-printed circles on a high quality filter paper (Whatman No.3) device, developed in-house (Figure 1).
The DBS were allowed to dry, at least for 2 hours at room temperature and then placed in individual zip lock plastic bags with a desiccant and stored at room temperature (28°C), for one week before testing. And, in case testing was delayed, the DBS were stored at 2-8°C until further testing for HCV RNA, maximum storage being for 1 year after collection [15]. The relative humidity was around 60%. Prior to further processing, the stored plasma samples and the DBS were thawed to room temperature.
HCV seroconversion panel PHV 920 Genotype 1a and HCV seroconversion panel PHV914 Genotype 2b (Seracare Life Sciences, BBI Diagnostics, USA) standards were taken as positive control in all the assays.
Molecular assays qualitative reverse transcription nested PCR [16] and a quantitative Real Time-PCR (Abbott Molecular Inc, USA) and Real Time-LAMP-developed in house, targeting the 5’ UTR gene of HCV were performed on the Plasma and the DBS from both the study and control groups.
RNA extraction from plasma (Qiagen, Germany): Using the QIAamp viral RNA mini kit (Qiagen, Germany), RNA was extracted from 140 µl of plasma according to the manufacturer’s instructions. Viral RNA was extracted from 140 μl of serum samples using QIAamp viral RNA mini kit (Qiagen, Germany) in accordance with the manufacturer’s instructions.
Figure 1: In house developed Filter paper device for collecting Dried Blood Spot.
Briefly, the sample was first lysed (lysis buffer) under highly denaturing conditions to inactivate RNases and to ensure isolation of intact viral RNA. Buffering conditions were then adjusted to provide optimum binding of the RNA to the QIAamp membrane, and the sample was loaded onto the QIAamp Mini spin column. RNA bound to the membrane, and other contaminants were efficiently washed away in two steps using two different wash buffers. Finally 50 μl of high-quality RNA was eluted in a special RNase-free buffer, ready for direct use or safe storage at -20°C.
RNA extraction from DBS (Qiagen, Germany) elution: From the DBS, two 6 mm spots were punched out and incubated in 2 ml lysis buffer (Qiagen, Germany) at room temperature for one hour. This lysate was used to extract the viral RNA using QIAamp viral RNA mini kit (Qiagen, Germany) as described above for the plasma.
cDNA synthesis: The complementary DNA (cDNA) was synthesized from 2 µl of extracted RNA in a 10 μl reaction volume with reverse transcriptase (RT) mix comprising of 5X-RT buffer, dNTPs, RNasin® ribonuclease inhibitor and Avian Myeloblast virus RT (AMV-RT) (New England Biolabs, USA).
Reverse Transcriptase Nested PCR was performed as described by Ponamgi et al. targeting the 5` UTR region of HCV [16].
First Round nested PCR: The 5’ non-coding region of HCV was amplified by adding 5 μl of cDNA prepared from RNA extracted from Plasma and corresponding DBS samples to the PCR master mix (New England Biolabs) containing the primers -
F1-5’-ACTGTCTTCACGCAGAAAGCGTCTAGCCAT-3’&
R1-5’- CGAGACCTCCCGGGGCACTCGCAAGCACCC-3’. A final volume of 25 μl was used. A no template control (NTC, reaction mix), a known HCV RNA (positive) control and water blank were also processed similarly for quality control and to exclude false-positive results in the PCR due to cross contamination. Thermal cycling was done on a programmable thermal cycler (Perkin -Elmer, USA) at 95°C for 2 min, 35 cycles at 94°C for 30 s, 50°C for 45 s, 72°C for 1 min and a final extension at 72°C for 5 min.
Second round nested PCR: 5 µl of the first-round PCR product was reamplified with internal primers F2 -5’-ACGCAGAAAGCGTCTAGCCATGGCGTTAGT-3’ and R2-5’-CCCGGGGCACTCGCAAGCACCCTATCAGG-3’ for another 35 cycles under the above conditions. PCR products were analyzed on 2% agarose gels with ethidium bromide and visualized in a gel documentation system (Syngene, UK). A 100-bp ladder (Fermentas) was used as a molecular weight marker.
HCV sequence specific primers were designed from the 5’ UTR region ranging from 99-314 (216 bp) target sequence of the HCV genome. The nucleotide sequence of the 5’ UTR gene of HCV was retrieved from Gen Bank (GenBank accession no. AY051292) and was aligned with the available 5’UTR gene sequences from global HCV strains including the circulating strains in India, to identify the conserved regions using DNASIS software (Hitachi, Japan).
The design of HCV specific LAMP outer forward primer (F3), outer backward primer (B3), forward inner primer (FIP) and backward inner primer (BIP) was done with the help of the Primer Explorer V4 software (http:/primerexplorer.jp/lamp) [14], An additional set of two loop primers (loop F and loop B) were designed to accelerate the amplification reaction. The primers were selected based on specific criteria [14].
All the primers were assessed for specificity before use in LAMP assays with BLAST search with sequences in Gen Bank (Table 1).
The RNA extracts from the plasma and the corresponding DBS and standard HCV RNA (positive) control were used for the HCV RT- LAMP. A negative control (reaction mix, NTC), was also processed similarly for quality control and to exclude false results in the RT- LAMP.
The RT-LAMP reaction was carried out in a 25 μl reaction mixture (Loop-amp RNA Amplification Kit; Eiken chemical co.Ltd., Tochigi, Japan) with the following reagents (final concentration): 20 mM TrisHCl (pH = 8.8),10 mM KCl,10 mM (NH4 )2 SO4 , 0.1% Tween20, 0.8 M betaine, 8 mM MgSO4, 1.4 mM dNTP each , enzyme mix containing 8 U of Bst DNA polymerase and 50 U of AMV reverse transcriptase each (Eiken Chemical Co.). The amount of primers needed for one reaction was: 40 pmol FIP and BIP, 20 pmol LF and LB, 5 pmol F3 and B3. Finally, an appropriate amount of template RNA was added to the reaction tube. The reaction was carried out in a water bath at 63ºC for 45 minutes and inactivated at 80ºC for 2 minutes.
Agarose gel analysis: Following incubation at 63°C for 45 min, a 10 µl of aliquot of the RT-LAMP assay product was electrophoresed on 2% agarose gel (Sigma Aldrich, USA) with ethidium bromide in tris acetate buffer and the results were documented by a gel documentation system (Syngene, UK).
Visual detection: Following amplification by RT-LAMP 0.2 μl of 1/10000 DMSO SYBR® Green I (SIGMA) was added to 25 μl of LAMP product. Any reaction tube which showed an observable color change from an initial orange to an apple green fluorescence (visualized with naked eye or under UV irradiation) was considered positive for HCV RNA, while a retained orange color was considered as a negative result.
The RNA extracts from the plasma and the corresponding DBS were tested with the HCV Real Time PCR kit on the ABI 7500 system (Life technologies, USA) using the F3 / B3 primers for evaluating the specificity of the primers. PCR cycles were programmed as follows: 50°C for 10 minutes for reverse transcription, 95°C for 5 minutes initial denaturation followed by 40 cycles of 95°C 30 seconds, 55°C for minutes and 72°C for 30 seconds. Melt curve analysis was programmed as follows: 95°C for 1 minutes, 55°C for 30 seconds and 95°C for 30 seconds by using SYBR Green Real time Master Mix -12.5 µl, RT/Taq mix -0.25 µl, Primer forward (F3) -0.125 µl, Primer reverse (B3) -0.125 µl, Nuclease free water- 10 µl, Template RNA- 2 µl extracted from Plasma and the corresponding DBS.

Table 1: Primer sequences used in HCV RT- LAMP targeting 5’ UTR of HCV Genome 216 bp Target, Genome position 99-314
To further evaluate the performance of the HCV RT-LAMP, the FDA approved Real Time HCV PCR (Real Time TM HCV Amplification reagent kit, Abbott Molecular Inc, USA) assay was performed for an automated reverse transcription, PCR amplification and detection/quantitation.
The assay was done by adding 50 µl of master mix (HCV oligonucleotide reagent including primers and probes, thermo stable rTth polymerase enzyme, and activation reagent) and 50 µl of extracted HCV RNA from both Plasma and corresponding DBS.
The cycling conditions were programmed as follows: Reverse Transcription at 59°C for 30 min, 4 cycles of Low stringent PCR - 95°C for 40 sec and 46°C for 30 sec, 6 cycles of High stringent PCR -92°C for 30 Sec and 60°C for 30 sec, 37 cycles of PCR amplification -92°C for 30 sec and 56°C for 30 sec followed by hold at 35°C for 40 seconds.
The LOD of HCV RT-LAMP was determined using HCV sero conversion panel PHV 920 Genotype 1a (Seracare Lifesciences, BBI Diagnostics, USA).
HCV RNA detection ability and stability in DBS were evaluated using the standard sample with a viral load of 5 x 105 copies / ml which was diluted in HCV RNA negative citrated whole blood to obtain dilutions of 10,100,1000, 10,000 and 1,00,000 copies/ml. DBS were prepared by spotting 50 µl of each dilution on the filter paper device, filling the spot and ensuring that the blood fully saturated through the filter paper. The cards were then dried at room temperature for at least 2 hours. The DBS were stored at 4°C until tested, for HCV RNA, by Real Time PCR and RTLAMP, using the procedures as described above.
The specificity of the HCV RT-PCR and RT-LAMP was determined by using NAs extracted from plasma of patients with other viral infections – 5 cases each of Hepatitis B (DNA), Hepatitis A and Chikungunya virus (RNA).
Inter run assay: Inter-assay variability for reproducibility, was assessed by testing 20 DBS elutes by HCV RT-LAMP on 3 days.
Intra run assay: The intra-assay variability for repeatability was assessed by simultaneously testing a single DBS spot elute, 10 times, by the HCV RT LAMP, on the same day.
The RT- LAMP products from 3 HCV RNA positive DBS that were negative for HCV RNA in the plasma were further sequenced for confirmation and to rule out false positive on DBS. The Nucleotide sequencing of the 5’UTR gene of HCV was carried out by employing the Big Dye Terminator Cycle Sequencing Ready Reaction kit with an ABI 3100 sequencer (Applied Biosystems, USA) by following the standard protocol [17]. The sequences were initially subjected to BLAST to find the closest sequence identity.
The male to female ratio among the study and control group was 4:1 with an age range of 40 to 60 years.
Detection of a 256 bp amplified product by HCV Reverse Transcriptase Nested PCR indicated that the DBS samples were positive for HCV RNA (Figure 2).

Figure 2: Nested RT-PCR for HCV. Well 1- 100 bp Marker (Fermentas, USA), Well 2 – NTC, Well 3- sample 1 Plasma, Well 4- Sample 2 Plasma, Well 5-Sample 1 DBS ,Well 6- Sample 2 DBS.
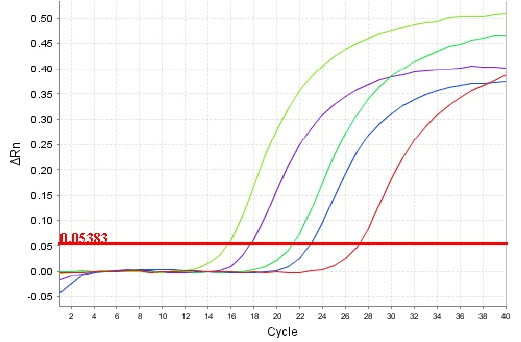
Figure 3: Real time amplification of HCV RNA by real time RT-PCR assay. Shown from left to right are the real time curves of HCV RNA amplification which rises in each cycle (X-axis).
DBS samples collected from HCV positive samples showed real time amplification by LAMP F3/B3 primers (Figure 3).
HCV specific primers were highly specific for the detection of HCV RNA with no cross reaction. HCV RT-LAMP primers did not amplify or cross reacted with any of the Hepatitis B (DNA), Hepatitis A and Chikungunya viral RNA template and 50 samples from healthy individuals, there by indicating their specificity. As depicted in (Figure 4), the size of the resultant product by RT-PCR using outer primers F3 and B3 was in good agreement with the predicted size (256 bp) for HCV RNA. 100% sequence homology was also observed between the primers and the corresponding nucleotide sequences. The specificity of the HCV RTLAMP was 100%, as none of the plasma and corresponding DBS samples with other viral infections were positive for HCV RNA by the HCV RTLAMP assay.
The RT-LAMP amplified products on the 2% Agarose gel were visualized as ladder-like bands, due to the formation of a mixture of stem-loop DNAs with various stem lengths. Results obtained with the visual detection by SYBR green method correlated with the agarose gel electrophoresis results.
The performance parameters of the 3 assays for the amplification of the target sequence of the 5’ UTR of HCV were correlating on both the specimen types (plasma and DBS). An acceptable linearity (r2 > 0.99) and intra and inter-run precision (CVs <10%) were recorded.
The Limit of detection for the HCV RT-LAMP assay and Real TimePCR were 50 copies /ml of HCV RNA (Figure 5).
All the 50 subjects in the control group were negative for HCV RNA by both HCV RT- LAMP and the real time PCR with both sample types. HCV RNA was also amplified from positive controls by both assays.
Using DBS, 145/300 (48.3%), (110/250 (44%) HD and 35/50 (70%) CLDs) were positive for HCV RNA by RT-PCR and RT-LAMP, while 3 of the DBS positive HD cases were negative with the corresponding plasma (107/250, 42.8%) by both methods. The RNA samples from the discrepant samples (3 DBS) were further amplified by RT-PCR by using F3 and B3 primers. The sequencing of this amplified product was carried out to confirm the results. The sensitivity of RT-LAMP from DBS is 100% and from plasma is 97.2%.
Sequencing of the RT- LAMP product from these 3 cases showed a 100% homology with the target sequence. The diagnostic performance of RT-LAMP assay from DBS against plasma is shown in (Table 2).
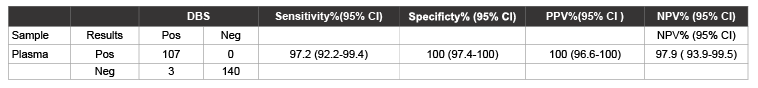
Table 2: Diagnostic performance of RT-LAMP assay from DBS against plasma from HCV patients in a tertiary care hospital in Hyderabad
The only limitation using the DBS was a prolonged elution step of one hour.

Figure 4: Agarose gel electrophoresis of HCV specific RT-PCR assay products on a 2% agarose gel employing F3 and B3 primers. Lane 1- 100 bp ladder (Fermantas, USA), Lane 2- NC, Lane 3- positive samples showing HCV specific product at 256 bp.
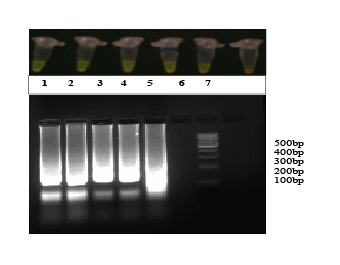
Figure 5: Limit of detection of HCV RT-LAMP. Lane 1-5 × 105 copies, Lane 2-5 × 104 copies, Lane 3-5 × 103 copies , Lane 4-5 × 102 copies , Lane 5-5 × 101 copies , Lane 6-5 × 100 copies of 5’UTR gene of Hepatitis C virus, Lane 7 - 100 bp ladder (Fermentas, USA)
Hepatitis C virus (HCV) infection remains frequent in patient receiving long-term dialysis both in developed and less-developed countries. There is abundant information on prevalence and incidence rates of HCV infection among patients on long-term dialysis in developed countries, and several population-based surveys have been made to this aim [18]. On the contrary, evidence on epidemiology of HCV in dialysis patients from developing countries is poor and mostly based on single-center studies [19].
Molecular techniques play a key role in the diagnosis and monitoring of treatment of HCV infection. Because it is difficult to culture HCV, molecular techniques were instrumental in first identifying HCV RNA, making it one of the first pathogens to be identified purely by molecular diagnostics [20].
With the recently improved molecular methods, the detection, monitoring and treatment of HCV infection represent a new paradigm in the field of virology, that is being increasingly addressed by the WHO and CDC, USA [7,21].
Detection of HCV RNA, the earliest marker of infection and a direct indicator of ongoing viral replication, is more reliable than serology in screening for an ongoing HCV infection especially in patients on dialysis and other immune compromised states, who may not mount an adequate antibody response. It also permits detection of infectivity during the seronegative window period, immediately after infection [6,22]. Therefore HCV RNA screening would be advisable for patients on hemodialysis, but a cost-effective analysis should be considered in this situation.
The HCV RT- LAMP developed in this study, targeted the 5`UTR region of the HCV genome, which was also used by other studies on HCV RTLAMP [11,14,23-25]. However, the earlier studies used serum specimen from known HCV positives, unlike in our study where the plasma and DBS were tested from study subjects who were high risk patients.
The HCV RT-LAMP assay was found to be a highly sensitive qualitative diagnostic test that is simple and easy to perform with minimum infra structure. The HCV RT-LAMP had a limit of detection (LOD) of 50 copies/mL similar to the nested RT- PCR and the HCV Real Time PCR assays. The LODs in other studies ranged between 8 to 84 copies of RNA / ml [11,14,23-25]. Usefulness of HCV RT-LAMP in the diagnosis of HCV RNA, especially in the early clinical diagnosis of acute HCV infection has been reported in earlier studies and it was found that there is no obvious difference in sensitivity between different HCV genotypes based on the RT-LAMP assay (11). However recent studies have shown the application of RT-LAMP assay for detection and genotyping of HCV in blood components with a sensitivity of 91.5% and specificity of 100% [26].
The HCV RT-LAMP assay has been appropriately evaluated and shown to meet defined performance targets and operational criteria in different kinds of patients, fulfilling all the criteria on diagnostic accuracy as per the Standards for Reporting of Diagnostic Accuracy (STARD) initiative [27]. The inter and intra test run parameters and the test evaluations indicate that the physical format of the test would not be influenced by local diagnostic practices and skills.
Since the LAMP assay does not require any sophisticated equipment and facility, except a water bath, it has a great potential to be applied as a simple tool not only for the rapid detection and diagnosis of HCV infection and monitoring response to therapy, but also can be used for surveillance and large scale epidemiological studies to assess the burden of HCV infection at remote and outreach areas [23].
Another major advantage of RT-LAMP is its rapidity requiring only 1.5 hours (including the extraction step) to perform the assay, compared to 2.5-3 hours for the real-time PCR assay. The flexibility of its detection method by visual fluorescence using sybr green is also a major advantage without the need for gel electrophoresis. Also, the calculated cost per test for the HCV RT-LAMP is 5 times less than the available HCV Real Time PCR and hence is affordable by any laboratory.
HCV RNA was stable at RT and 2-8°C for up to 12 weeks, which should allow for convenient shipment and storage. DBS is an effective method for HCV screening in patients on haemodialysis with limited venous access and in patient care in resource poor settings.
This study has highlighted the use of DBS in the Indian climatic conditions, storage of longer durations and for large-scale surveys using serological assays. To the best of our knowledge, the performance characters of the HCV RT- LAMP on DBS specimen were evaluated for the first time in India, through this study. As shown in the results, 3 of the plasma samples from HD cases were negative for HCV RNA, probably due to the loss of the RNA in the plasma. Sequencing of the RT-LAMP product from these 3 DBS HCV RNA positive cases showed a 100% homology with the target sequence. Using the DBS as the source of the blood sample, the robust performance parameters of the HCV RT- LAMP support the utility of the assay as an important screening tool that can be applied at remote settings both for detection as well as surveillance purposes. Except that the elution step prolonged the test duration by an hour, the DBS method is a convenient and reliable procedure for blood sample collection. It is robust and less prone to deterioration and contamination even on prolonged storage unlike the plasma specimen [28].
The authors would like to thank DRDO, Delhi and the director DRDE, Gwalior for funding this study.
The authors declare that there is no conflict of interests regarding the Publication of this article.
Download Provisional PDF Here
Article Type: Research Article
Citation: Lakshmi V, Neeraja M, Lavanya V, Priyanka EN, Sharma S, et al. (2016) Application of Real Time Loop Mediated Isothermal Amplification Assay on Dried Blood Spots in the Detection Of HCV RNA among High Risk Patients. J Emerg Dis Virol 2(1): doi http://dx.doi.org/10.16966/2473-1846.111
Copyright: © 2016 Lakshmi V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access