Introduction
Due to their capacity for self-renewal and their potential to
differentiate into all blood cell lineages, hematopoietic stem cells (HSCs)
are considered to be the exclusive source of the hematopoietic system [1-
5]. Although HSCs can be used to treat blood disorders and malignant
diseases [6], their clinical application has been limited, primarily because
HSCs represent only a small percentage of bone marrow (BM) cells and
because it is difficult to maintain their self-renewal capacity in vitro and to
avoid the immune rejection of HSCs during transplantation [2,3].
Induced pluripotent stem (iPS) cells, which can be produced via the
over expression of specific factors [7-9], exhibit the capacity for selfrenewal
and the potential to differentiate into any cell type; therefore, these
cells are of great interest for disease treatment [10,11], drug screening
[12], toxicology and regenerative medicine [13]. However, the generation
of iPS cells is time-consuming and inefficient (0.001-0.1%) and, most
importantly, involves several safety concerns [14,15]. To overcome these
issues, investigators have developed methods for the direct conversion of
cells [16-18]. According to recent reports, these reprogramming processes
generate a variety of intermediate cell types; for instance, iPS cells are one
of the products of somatic reprogramming. However, the final results
of these reprogramming processes depend on many specific conditions
[18,19]. Bhatia’s group reported that human fibroblasts could be converted
into multi-lineage blood progenitors via the binding of transcription
factors (TFs) to hematopoietic regulatory regions [19], and their results
indicate that Oct4 acts as a lineage-specific TF [18].
In addition, it was recently reported that a combination of four
transcription factors, Gata2, Gfi1b, c-Fos and Etv6, induces hematopoietic
cell formation from endothelial-like precursor cells. These cells exhibit a
CD45+cKit+CD150+CD48-
phenotype that is similar to that of LT-HSCs
[20]. However, the hematopoietic potential of these reprogrammed cells
is limited. Another report showed that the combination of Run1t1, Hlf,
Lmo2, Prdm5, Pbx1 and Zfp37 promotes the conversion of blood cells
to HSCs, suggesting that specific factors regulate the gene networks of
HSCs [21]. However, as blood cells are a distinct cell type, the conversion
of MEF cells to HSCs remains a challenge. More recently, the expression
of FOSB, GFI1, RUNX1 and SPI1 in human umbilical vein endothelial
cells and human adult dermal microvascular endothelial cells was found
to induce the generation of hematopoietic cells displaying long-term MPP
activity [22], although obtaining human endothelial cells is difficult. In
contrast, MEFs are easily obtainable for use in reprogramming processes.
Lacaud’s group also demonstrates that hematopoietic transcription factors
induced conversion of fibroblasts to hematopoietic progenitors. However
long-term in vivo engraftment capacity was not shown [23].
An in-depth understanding of the hematopoietic system and recent
findings on somatic cell reprogramming motivated us to examine
whether mouse somatic cells can be reprogrammed into hematopoietic
progenitor cells (HPCs) using a combination of HSC-specific factors
and pluripotency-related genes and then culturing these transduced
cells under HSC-specific culture conditions. In this study, we were
able to generate lineage-c-Kit+Sca-1+ (LKS), long-term HSC (LT-HSC;
CD34- Flk2- CD150+CD48- LKS), short-term HSC (ST-HSC; CD34+Flk2- CD150+CD48- LKS) and MPP (CD34+Flk2+CD150- CD48- LKS) cell populations [24,25] by transfecting mouse embryonic fibroblasts (MEFs) with pMXs-Oct4/Sox2/Klf4/c-Myc/Lmo2/c-Fos/c-Myb (pMXs-7TF MEFs). Furthermore, pMXs-7TF MEFs were successfully differentiated
into immune cells both in vitro and in vivo. It is highly likely that the
successful generation of HPCs will resolve the primary difficulties
associated with the use of matched HSCs in individual patients and with
the in vitro expansion of HSCs for clinical use.
Materials and Methods
Mice
C57BL/6J mice and congenic CD45.1+ C57BL/6 mice were purchased
from The Jackson Laboratory (Bar Harbor, ME, http://www.jax.org), and
Rag2-/-II2γg-/- mice (C57BL/6J X C57BL/10SgSnAi) were purchased
from Taconic Farms [26]. The hCD34 transgenic mice (C57B6) carrying
a PAC clone containing the entire hCD34 gene and 12.8 kb of 5’ and 25.6
kb of 3’ flanking sequences were constructed as previously reported [27].
All mice were maintained under specific pathogen-free conditions, and
we used 8 to 12-week-old male mice. All experiments were conducted
in accordance with the guidelines of the Korea Research Institute of
Bioscience and Biotechnology (KRIBB) Institutional Animal Care and
Use Committee (KRIBB-AEC-13013).
Cell culture
HSCs (Lin-c-Kit+Sca-1+; LSK) or Lin-c-Kit+ (LK) cells were isolated
from mouse BM using a FACSAria flow cytometer (BD Biosciences).
pMXs-7TF MEFs and HSCs were cultured in Myelocult long-term
culture medium (ST05350, STEMCELL Technologies, Vancouver, British
Columbia, Canada) supplemented with 2 µg/ml indomethacin, 20 µg/
ml gentamicin, 30 ng/ml SCF, 50 ng/ml Flt3L, 20 ng/ml TPO and 20 ng/
ml IL-6. All cytokines were purchased from PeproTech (Rocky Hill, NJ,
USA). MEF cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) containing 10% fetal bovine serum (HyClone).
MEF preparation
MEFs were prepared from 13.5-day pregnant mice and were used for
1-2 passages. The heads and internal organs of the mice were removed,
and the torsos were minced and dispersed in 0.1% trypsin (HyClone)
for 30 min at 37°C. The cells were cultured for two population doublings
(considered as one passage) and then viably frozen. These MEFs were used
for all subsequent experiments. The MEFs were maintained in DMEM
containing 10% fetal bovine serum (HyClone) and were sub cultured at
a dilution of 1:3 upon reaching confluence. MEFs cultured for fewer than
three passages were used for pMXs-7TF MEFs generation and feeder cell
preparation.
Retroviral infection
Oct 3/4, Sox2, Klf4, c-Myc, LIM domain only 2 (Lmo2), c-Fos,
c-myeloblastosis (c-Myb) and c-Jun were transduced using the pMXs
vector. The pMXs plasmids expressing Oct 3/4, Sox2, Klf4 and c-Myc were
obtained from Addgene. The Lmo2, c-Fos, c-Myb and c-Jun fragments
were obtained from the 21C Frontier Human Gene Bank (KRIBB). To
generate pMXs-expressing Lmo2, c-Fos, c-Myb and c-Jun plasmids,
c-Fos, c-Myb and c-Jun were introduced into the BamHI/HindIII sites
of the pMXs plasmid; Lmo2 was introduced into the BglII/HindIII sites
of pMXs. Platinum-E retroviral packaging cells (Plat-E cells, Cell Biolabs
Inc., [28]) were seeded on six-well plates at 6 × 105
cells per well. The
next day, these cells were transfected with the pMXs retroviral vectors
using Lipofectamine and PLUS reagents (Invitrogen) according to the
manufacturer’s instructions. After four hours, the medium was replaced
with 5 ml DMEM containing 10% FBS, and the cells were incubated at
37°C in an incubator. After 48 hours, the medium was collected as the
first virus-containing supernatant and was replaced with fresh medium.
After 24 hours, the second virus-containing supernatant was collected.
The viruses were filtered using a 0.45-μm cellulose filter (Millipore), and
equal amounts of virus were mixed and transferred to the MEFs in the
presence of 4 g/ml Polybrene (Sigma-Aldrich). This infection process was
repeated every 12 hours for a total of three times.
Induction of HPCs
To generate HPCs, MEFs were co-infected with pMXs-Oct 4/Sox2/
Klf4/c-Myc/Lmo2/c-For/c-Myb for 3 days. The day after infection, the
cells were reseeded at 1–2 × 105
cells per well in 0.1% gelatin-coated sixwell
plates containing Mitomycin C (Sigma-Aldrich)-treated MEF feeder
cells. The cells were cultured in Myelocult long-term culture medium
(ST05350, STEMCELL Technologies, Vancouver, British Columbia,
Canada) supplemented with HSC cytokines (2 µg/ml indomethacin, 20
µg/ml gentamicin, 30 ng/ml SCF, 50 ng/ml Flt3L, 20 ng/ml TPO and
20 ng/ml IL-6). After 14-21 days, colonies were picked using a 100 µl
pipette. To break the colonies into small mass, the colonies were treated
with trypsin-EDTA (Hyclone SH30042.01) for 15min at 37°C. The
colonies were reseeded in MEF feeder cells. The colonies were cultured
in Myelocult long-term culture medium supplemented with HSC
cytokines. To ensure the generation of HPCs, the colonies were stained
for HSC markers (c-Kit+, Sca-1+, CD34+/-, Flk2+/-, CD150+/-, CD48+/-, and
lack lineage markers) and were analyzed via FACS. To confirm HPCs
generation, we performed PCR and western blot analysis. To validate the
retrovirus-mediated over expression of the candidate genes, the mRNA
levels of each candidate gene and the GAPDH gene were determined via
RT-PCR analysis.
Flow cytometry
To analyze HSC marker expression, the cells were stained with
antibodies against CD34, FLK2, hCD34, CD48, CD150, CD117 and
Ly6a for 30 min at 4°C. Lineage staining was performed using a mixture
of biotinylated anti-mouse antibodies against Mac-1 (CD11b), Gr-1
(Ly6C), Ter-119, NK1.1, CD2 and B220. To distinguish between donor
and recipient cells, antibodies against CD45.1 and CD45.2 were used,
the donor cells were stained using the CD45.2 antibody and the recipient
cells were stained using the CD45.1 antibody. To analyze myeloid and
erythroid marker expression, antibodies against Gr-1, Mac-1 and Ter-
119 were used. These antibodies were purchased from BD Biosciences or
eBiosciences. The data were generated using a FACSCanto flow cytometer
(BD Biosciences) and were analyzed using FlowJo software (ThreeStar,
Ashland, OR, USA).
Western blot analysis
Cells were lysed in lysis buffer containing 1 mM EDTA, 1 mM
EGTA, 20 mM Tris-HCl, 10 mM NaCl, 1% Triton X-100, 30 mM
sodium pyrophosphate, 25 mM β-glycerol phosphate, 1 mM Na3
VO4
and 1 mM PMSF. The lysates were resolved via sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE), and the separated
proteins were transferred to PVDF membranes (Millipore) and stained
with 0.1% Ponceau S solution. After blocking with 5% nonfat milk or BSA,
the membranes were incubated in anti-c-Kit (Abcam, Cambridge, MA,
USA), anti-CD150 (Abcam, Cambridge, MA, USA) or anti-actin (Santa
Cruz Biotechnologies, Santa Cruz, CA, USA) antibodies.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
and Quantitative RT-PCR (QPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen). For the
RT-PCR analyses, each RNA was reverse-transcribed into cDNA using
M-MLV reverse transcriptase and oligo (dT) 15 primers (Promega,
Madison, WI, USA). Then, this cDNA was amplified using Emerald Amp
PCR Master Mix (Takara), and qPCR was performed using SYBR Premix
Ex Taq (Takara) according to the manufacturer’s protocol. All RT-PCR
and qPCR data were normalized to the GAPDH expression levels. The
primer sequences used are listed in Supplementary table 1.
Colony-Forming cell (CFC) assays
To perform the CFC assays, Methocult M3434 medium (STEMCELL
Technologies) was used. First, 0.4 ml of pMXs-7TF MEFs was added
to 4 ml of methylcellulose-based medium containing the appropriate
cytokines, and the tubes were vortexed to ensure the mixing of the cells
with the medium. The cell mixtures were incubated for 5 min; then, 1.1 ml
of the mixtures was inserted as droplets into a 30-mm dish. The cells were
incubated at 37°C in 5% CO2
at 95% humidity for 14 days. The colonies
were morphologically evaluated and counted after 3, 7 or 14 days using an
inverted microscope at 40X magnification.
Giemsa staining
To further analyze differentiation and proliferation, the cells were
collected and resuspended in cold PBS. Then, the cells were transferred to
glass slides using a Cytospin centrifuge for Giemsa staining. The cells were
fixed with methanol for 2 min and stained with Giemsa staining solution
(Sigma) for 4 min. The slides were transferred to phosphate buffer and
rinsed with water. After the slides were completely dry, mounting
solution was applied as droplets, and the slides were coverslipped.
Photomicrographs were captured at 1000X magnification.
in vitro differentiation of NK cells from pMXs-7TF MEFs
NK cell differentiation from the HSCs was performed essentially as
described previously [29]. In brief, HSCs or pMXs-7TF MEFs were seeded
in 12-well plates and cultured in RPMI 1640 medium containing 10% heatinactivated
FBS, indomethacin (2 µg/ml, Sigma), gentamicin (20 µg/ml),
SCF (30 ng/ml, PeproTech), Flt3L (50 ng/ml, PeproTech) and IL-7 (0.5 ng/
ml, PeproTech) for seven days to generate precursor natural killer (pNK)
cells. The pNK cells were then harvested, reseeded and cultured in the
presence of IL-15 (50 ng/ml, PeproTech) for six days to produce mature
natural killer (mNK) cells. The purity of the mNK cells was determined
via FACS analysis using anti-CD3e and anti-NK1.1 antibodies.
in vitro differentiation of the Myeloid lineage from pMXs-7TF
MEFs
To differentiate cells of the myeloid lineage, pMXs-7TF MEFs and
MEFs transfected with the pMXs control vector were seeded in 12-well
plates and cultured in RPMI 1640 medium supplemented with 10 ng/ml
IL6, 10 ng/ml GM-CSF, 10 ng/ml G-CSF, 50 ng/ml SCF, 20 ng/ml IL3 and
10 ng/ml BMP4. After two weeks, we observed that the pMXs-7TF MEFs
were able to differentiate into the myeloid lineage. To analyze myeloid
marker expression via FACS, the cells were stained with anti-Mac-1 and
anti-Gr-1 antibodies.
Cytotoxicity analysis
NK cell death was evaluated using a calcein-AM release assay
(Invitrogen) as previously described [29]. In brief, Yac-1 cells were
incubated in 5 µg/ml calcein-AM for 1 hour at 37°C with occasional
shaking. After washing, the cells were plated at the indicated effector:target
cell (E:T) ratio in 96-well round-bottom plates and were incubated for
four hours at 37°C. Following incubation, 100 µl of the supernatant was
collected, and the plates were analyzed using a fluorescence plate reader
(excitation filter 485 nm, emission filter 530 nm, BerThold, Germany).
Reconstitution assay
For the competitive reconstitution assays, MEF (CD 45.2+, 5 × 104
) or
LK cells (CD45.2+, 5 × 104
) from pMXs-7TF MEFs or BM cells were mixed
with whole BM cells (1 × 106
) from C57BL/6 (CD 45.1+) mice. Then, these
mixtures were intravenously injected into eight-week-old C57BL/6 CD
45.1+ congenic mice that had received lethal γ-irradiation (9 Gy). For
serial BM transplantation (BMT), donor-derived BM cells (3 × 106
) from
recipients were injected into a second set of recipient mice (CD 45.1+) at
16th week after the first BMT. Peripheral blood was collected and analyzed
via FACS to determine the repopulation percentage of the donor-derived
cells. Successful pMXs-7TF MEFs transplantation was confirmed based
on the identification of donor-derived HSC-positive cells in the BM at
16th week after second transplantation. These transplanted recipients were
maintained under specific pathogen-free conditions.
Statistical analysis
The data are presented as the mean ± SD values, unless stated otherwise.
To compare two groups, we performed two-tailed paired t-tests using
PRISM software (San Diego. CA, USA) and performed two-tailed paired
Student t-tests using Microsoft Excel. A p value of less than 0.05 was
considered to be significant.
Results
Reprogramming MEFs to generate HPCs
To generate HPCs, we selected candidate genes based on the results
from an earlier serial analysis of gene expression (SAGE) dataset
(Supplementary figure 1A) [30]. According to the SAGE dataset, c-Jun
(J), Lmo2 (L), c-Fos (F), c-Myb (B), and c-Myc (M) were higher expressed
in HSCs than natural killer (NK) cells. These TFs are known to play a
role in the development of lymphoid lineages. To confirm the increased
expression of these TFs in HSCs (LKS cells), we verified the mRNA
levels of these candidate genes in HSCs via RT-PCR analysis. As shown
in supplementary figure 1B, these genes displayed increased expression
levels in HSCs compared with other cell types, such as MEFs and pNK,
mNK cells and embryonic stem (ES) cells. Next, these candidate genes
were applied to the MEFs in combination with pluripotency-related
genes, including Oct4, Sox2, Klf4 and c-Myc (OSKM), using the pMXsbased
retrovirus system. Flow cytometric analysis showed that RFPpositive
MEF cells comprised more than 90% of the cells (Supplementary
figure 1C) and that each gene was specifically overexpressed in the MEFs
(Supplementary figure 1D).
For reprogramming, MEFs were transduced with the candidate
genes and cultured on MEF feeder cells in long-term HSC culture
medium (Myelocult, M5300) lacking leukemia inhibitory factor (LIF),
a compound that plays a crucial role in the maintenance of self-renewal
in mouse ES cells [31]. After 14-21 days in culture, colonies were picked
and were seeded on MEF feeder cells in long-term HSC culture medium
supplemented with indomethacin, gentamicin, and hematopoietic
cytokines, such as SCF, Flt3L, TPO and IL-6. To ensure that we generated
HPCs, the colonies were stained for HSC markers and were analyzed
via FACS. The general scheme is depicted in figure 1A. The various
combinations of candidate genes were tested to determine the optimal
genes for transduction (Supplementary figure 2A). The number of
colonies depended on which of the genes were combined with OSKM. The
combinations, such as OSKML, OSKMF, OSKMB, OSKMLF, OSKMLB,
OSKMFB, and OSKMLFB induced colony formation (Supplementary
figure 2B). In particular, the OSKMLFB combination produced the largest
number of colonies (Supplementary figure 2C) and generated a distinct
population of LKS cells (Supplementary figure 2D). Therefore, we selected
the OSKMLFB combination (pMXs-7TF) as the optimal reprogramming
conditions. figure 1B shows the morphology of control MEFs (pMXs),
pMXs-7TF MEFs, and primitive HSCs. The pMXs-7TF MEFs exhibited
a round morphology that was similar to that of primitive HSCs.
Transduction with pMXs-7TF increased the number of formed colonies
compared to transduction with pMXs (Figure 1C). Next, to define the
emergent hematopoietic cells, we examined the time-course of gene
expression during pMXs-7TF MEFs generation. After transduction, the
cells were harvested at the indicated times, and HSC marker expression
was analyzed via qPCR. The expression of fibroblast-specific genes, such
as Vim and Acta2, decreased gradually (Figure 1D and supplementary
figure 3A), but the expression of HSC-specific markers, including c-Kit,
Sca-1, CD150, CD45, Il3ra and Csf3r [20], increased in cells transduced
with pMXs-7TF (Figure 1E and supplementary figure 3B). Western blot
analysis confirmed that c-Kit and CD150 were highly expressed in the
pMXs-7TF MEFs compared with the control pMXs-transduced cells
(Figure 1F).
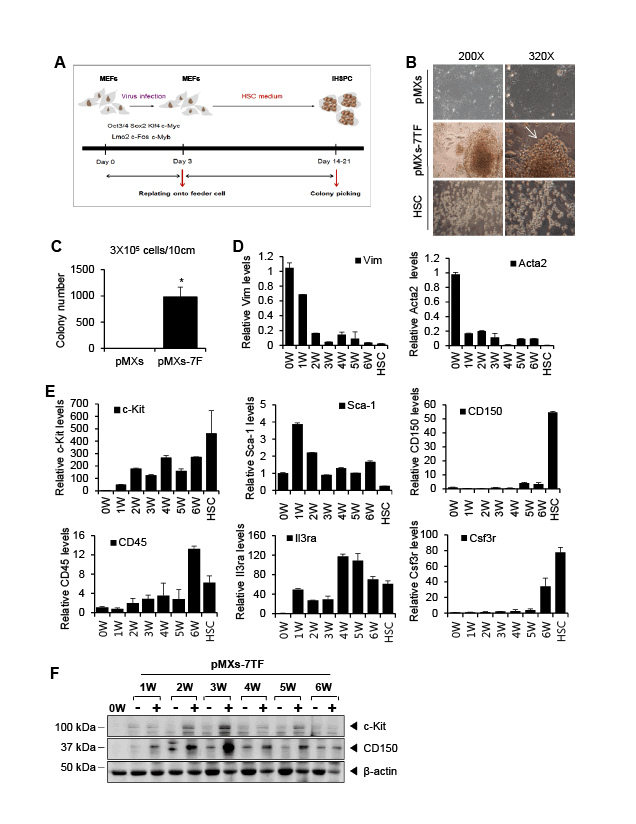
Figure 1: Reprogramming of MEFs to HPCSs using seven factors. (A) A schematic showing the pMXs-7TF MEFs reprogramming process. MEFs
were infected with retrovirus for a total of three days. After the first day, the virus-infected MEFs were reseeded on Mitomycin C-treated MEF feeder
cells. The cells were cultured in Myelocult long-term medium supplemented with several hematopoietic cytokines. After 14-21 days, colonies were
picked and reseeded in 24-well plates. (B) A bright-field micrograph showing the morphology of the cells transduced with the pMXs control vector or
the pMXs-7F vector and HSCs. Small round cells were detected among the pMXs-7F-treated cells (white arrow). Original magnifications: 200X and
320X. (C) The MEFs were transduced with the seven transcription factor-expressing (pMXs-7TF) vector or the pMXs control vector. The number of
colonies was counted 14-21 days after infection. The error bars represent the s.d. (n=3); *p<0.05 compared with the pMXs-transduced MEFs. (D)
The relative expression levels of fibroblast-associated genes (Vim and Acta2) in the pMXs-7TF-transduced MEFs. The transduced MEFs or colonies
were harvested at 0-6 week (W) after reseeding on the MEF feeder cells. Target gene expression was evaluated via qPCR and was normalized to the
GAPDH expression levels. HSCs were used as controls. (E) MEFs were transduced with the seven transcription factor-expressing vector (pMXs-7TF).
After transduction, the cells were harvested at the indicated times, and HSC marker expression was analyzed. HSCs or ES cells were used as controls.
Target gene expression was evaluated via qPCR and was normalized to the GAPDH levels. All experiments were independently repeated at least
three times, and the error bars represent the s.d. (F) Time course analysis of HSC marker expression. The expression of c-Kit, CD150 and β-actin was
determined via Western blot analysis. (–) indicates pMXs-control vector-transduced cells; (+) indicates pMXs-7TF-transduced cells.
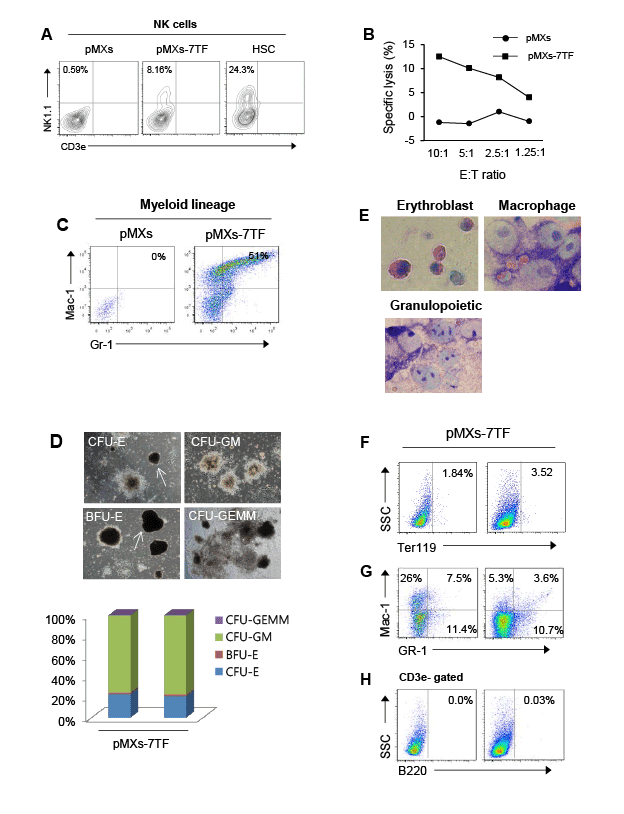
Figure 2: Multipotent capacity of pMXs-7TF MEFs in vitro. (A) To explore the capacity of the cells to differentiate into NK cells, the HSCs or the pMXs-
7TF MEFs were co-cultured with Mitomycin C-treated OP9 cells in the presence of Flt3L, SCF and IL-7 for 6 days. After 6 days, to generate mNK
cells, the cells were cultured in the presence of IL-15 for 6 additional days. Then, the cells were stained with anti-CD3 and anti-NK1.1 antibodies. The
phenotypes of the CD3e-NK1.1+ subsets were determined via FACS analysis. The percentage of NK cells among the gated lymphocyte population
was evaluated. HSCs were used as positive controls. (B) The cytotoxicity of mNK cells derived from pMXs-7TF MEFs or pMXs-transduced MEFs. The
mNK cells were incubated in calcein-AM-labeled Yac-1 cells at the indicated E:T ratio (10:1, 5:1, 2.5:1 or 1.25:1). After four hours, the supernatant was
collected, and the plate was examined using a fluorescence plate reader (485 nm excitation, 530 nm emission). All experiments were independently
repeated at least three times. (C) To induce the myeloid differentiation of pMXs-7TF MEFs in vitro, the pMXs-7TF MEFs were maintained in myeloid
culture medium containing several cytokines. After two weeks, the cells were stained with anti-Mac-1 and anti-Gr-1 antibodies and were analyzed via
flow cytometry. (D) CFC images and the relative frequency of CFU formation from the pMXs-7TF-transduced cells. The left panel shows images of
CFU-E, CFU-GM, BFU-E, and CFU-GEMM colonies. Original magnification: 40X. The right panel shows the relative frequency of colony formation.
Representative results from three independent experiments are shown. (E) Giemsa staining of Methocult colonies derived from pMXs-7TF MEFs on
day 14. Original magnification: 1000X. (F-H) Flow cytometry to determine the differentiation states of the cell populations. Erythroid cells (F), myeloid
cells (G) and B cells (H). After 14 days, Methocult colonies were harvested and stained with lineage-specific antibodies.
To validate the commitment to pluripotency, we examined the
expression patterns of pluripotency markers at the indicated time points.
The total levels of Oct4, Sox2 and Klf4 decreased in a time-dependent
manner, and the expression level of c-Myc, which is highly expressed in
HSCs, was elevated in the pMXs-7TF MEFs and HSCs (Supplementary
figure 4A). The endogenous expression of Oct4 and Sox2 was lower in the
pMXs-7TF MEFs than in the ES cells (Supplementary figure 4B). Nanog
and E-Ras, which are markers for pluripotent cells, were expressed at basal
levels in the pMXs-7TF MEFs (Supplementary figure 4C). Taken together;
these findings indicated that the pMXs-7TF MEFs reprogramming
process does not require the generation of iPS cells.
To further analyze the cellular phenotypes, we examined surface
antigen expression in the transduced cells. LKS cell, long-term HSC, shortterm
HSC and MPP (CD34+Flk2+CD150-
CD48-
LKS) cell populations
were detected in the pMXs-7TF MEFs that were derived from individual
colonies. As shown in supplementary figures 5A-5B, Sca-1, but not c-Kit,
was expressed in the MEFs transduced with the pMXs control vector.
Sca-1 is expressed in a variety of cells, including mesenchymal stem cells,
cardiomyocytes and fibroblasts [32,33]; this broad expression explains the
detection of Sca-1 in the pMXs-transduced cells. Although surface antigen
expression in the pMXs-7TF MEFs was not exactly the same as that in
the primitive HSCs, the pMXs-7TF MEFs clearly included an LKS cell
population. Next, we performed qPCR to compare the gene expression
profiles between pMXs-7TF MEFs, HSCs and ES cells (Supplementary
figure 5C). The expression levels of these HSC markers in the pMXs-7TF
MEFs were similar to those in the HSCs. Next, we performed RT-PCR
analysis. As shown in supplementary figure 5D, HSC surface markers,
such as c-Kit, Sca-1, CD150 and CD34, and TFs, such as c-Jun, Lmo2,
c-Fos, c-Myb and Trim28, that are highly expressed in HSCs were strongly
expressed in our pMXs-7TF MEFs and HSCs. Nanog was not expressed
in the pMXs-7TF-transduced cells. In addition, the protein levels of these
factors were verified via Western blot analysis. As expected, c-Kit and
CD150 were expressed in the pMXs-7TF MEFs (Supplementary figure 5E),
and these results were verified via immunocytochemistry using antibodies
against SSEA (a pluripotent cell marker), c-Kit and Sca-1 (Supplementary
figure 5F). The HSC surface markers c-Kit and Sca-1 were highly
expressed in pMXs-7TF MEFs and HSCs, but SSEA was not expressed in
pMXs-7TF MEFs. Taken together, these results demonstrated that pMXs-
7TF MEFs display similar molecular characteristics to primitive HSCs.
To determine whether these pMXs-7TF MEFs continually expand and
maintain their HSC properties in vitro, pMXs-7TF MEFs were cultured
in long-term HSC culture medium for two months. We found that the
LKS cells population remained present after two months of culturing
(Supplementary figure 5G).
Assessment of differentiation potential in vitro
Next, we examined the multipotency of the pMXs-7TF MEFs by
measuring their capacity to differentiate into the lymphoid and myeloid
lineages. To confirm lymphoid lineage differentiation, we generated NK
cells from pMXs-7TF MEFs using a two-step in vitro differentiation
protocol [34]. As shown in figure 2A, the pMXs-7TF MEFs could be
differentiated into CD3e-NK 1.1+ mNK cells, as has been observed for
primitive HSCs, and these cells were positive for other NK cell markers,
such as DX5 (Supplementary figure 6A) [35]. We then examined the
cytotoxicity of mNK cells derived from pMXs-7TF MEFs. The mNK
cells that were derived from pMXs-7TF MEFs (Supplementary figure 6B)
displayed cytotoxic activity against Yac-1 tumor cells, whereas the pMXstransduced
cells did not display this activity (Figure 2B). Next, to verify
the differentiation of pMXs-7TF MEFs into the myeloid lineage, the cells
were cultured in differentiation medium containing several cytokines,
including IL6, GM-CSF, G-CSF, SCF, IL3 and BMP4. After two weeks, we
observed that the pMXs-7TF MEFs differentiated into the Gr-1+Mac-1+
myeloid lineage (Figure 2C). To determine the specific hematopoietic
potential of the pMXs-7TF-transduced cells, we performed a CFC assay
using methylcellulose-based medium supplemented with recombinant
cytokines (M3434). Erythroid colony-forming unit (CFU-E), granulocytemacrophage
colony-forming unit (CFU-GM), erythroid burst-forming
unit (BFU-E) and granulocyte-erythroid-macrophage-megakaryocyte
colony-forming unit (CFU-GEMM) colonies were successfully derived
from the pMXs-7TF MEFs (Figure 2D). The colonies derived based
on the CFC assay were stained with Giemsa, and the majority of the
colony-forming cells exhibited granulocyte and macrophage lineage
characteristics (Figure 2E). To further verify the identity of these lineages,
the colonies derived based on the CFC assay were stained with antibodies
against Mac-1 and GR-1 (myeloid lineage, figure 2F), Ter119 (erythroid
lineage, figure 2G) and B220 (lymphoid lineage, figure 2H). We found
that these cells expressed myeloid and erythroid markers. These results
confirmed the colony-forming potential and multilineage differentiation
capacity of these pMXs-7TF-MEFs.
Functional properties of the pMXs-7TF MEFs in vivo
HSCs are crucial for the reconstitution of hematopoiesis upon
transplantation into recipients who have undergone BM ablation
[3,36,37]. To investigate the ability of these pMXs-7TF MEFs to regenerate
the hematopoietic system, we cultured the reprogrammed cells on MEF
feeder cells. After 2-3 weeks, LK cells from pMXs-7TF MEFs (CD45.2+)
or BM (HSC, CD45.2+) were sorted and transplanted with congenic
competitors (BM, CD45.1+) into lethally γ-irradiated congenic mice
(CD45.1+). After 16 weeks, we analyzed the reconstituted cell population
via FACS analysis (Figure 3A). To verify the engraftment of donor-derived
cells in the blood, we collected peripheral blood at 5, 9 and 12 weeks after
transplantation. Similar to BM-derived LK cells (HSC), pMXs-7TF MEFs
-derived LK cells (pMXs-7TF) exhibited the capacity for reconstitution in
the peripheral blood (Figures 3B-3C), and donor-derived CD45.2+ LKS
cell populations were detected in the BM of the transplant-recipient mice.
In addition, donor-derived (CD45.2+) immune cells, including NK cells,
T cells and B cells were observed in the recipient mice (Figure 3D and
supplementary figure 7A).
To confirm the reconstitutive properties of these cells, pMXs-7TF
MEFs were transplanted into Rag2/II2 g double-knockout mice, which
contain no detectable NK1.1+ NK cells or Thy1+ T cells [38]. Nine weeks
after transplantation, NK1.1+DX5+NK and CD4+CD8+ T cells were
observed in the spleens of the pMXs-7TF MEFs- and HSC-transplantrecipient
mice (Figure 3E and supplementary figure 7B). As previous
reports indicated that Oct 4 can induce the direct conversion of blood
progenitors from human fibroblasts and that these cells can differentiate
into a myeloid lineage but not lymphoid cells [19], we examined the
presence of myeloid-lineage cells in recipient mice (CD45.1) transplanted
with LK cells (CD45.2) derived from pMXs-7TF MEFs or BM cells. We
found that CD45.2+Gr-1+Mac-1+ myeloid lineage cells were present in
the recipient BM (Figure 3F and supplementary figure 7C). These data
demonstrated that pMXs-7TF MEFs exhibit the potential to differentiate
into both the lymphoid and myeloid lineages.
To confirm the ability of these pMXs-7TF MEFs to regenerate the
hematopoietic system, we performed a serial BMT. LK cells from
pMXs-7TF MEFs (CD45.2+) or BM (HSC, CD45.2+) were sorted and
transplanted with congenic competitors (BM, CD45.1+) into lethally
irradiated congenic mice (CD45.1+) At 16 week after the first BMT, donorderived
BM cells from recipients were injected into secondary recipient
mice (CD 45.1+) (Figure 4A). At 16 week after each BMT, peripheral blood
(PB) was collected and analyzed via FACS to determine the repopulation
percentage of the donor-derived cells (Figure 4B and supplementary figure
8A). At 16 week after second BMT, we analyzed the reconstituted cell
population via FACS analysis. First, to verify the engraftment of donorderived
cells in the blood, we collected PB at 4, 8, 12 and 16 weeks after
transplantation (Figure 4C and supplementary figure 8B). Similar to BMderived
LK cells (HSCs), pMXs-7TF-MEFs-derived LK cells exhibited the
multi-lineage reconstitution through the degree of B cells and myeloid
cells in the PB (Figures 4D-4E and supplementary figure 8C). In addition,
donor-derived CD45.2+ HSC populations were detected in the BM of
the transplant-recipient mice (Figure 4F and supplementary figure 9A).
Donor-derived (CD45.2+) immune cells, including NK cells, T cells, B
cells and Myeloid cells were observed in the recipient mice (Figure 4G,
supplementary figures 9B and 9C). These data demonstrated that pMXs-
7TF MEFs exhibit the potential to differentiate into both the lymphoid
and myeloid lineages.
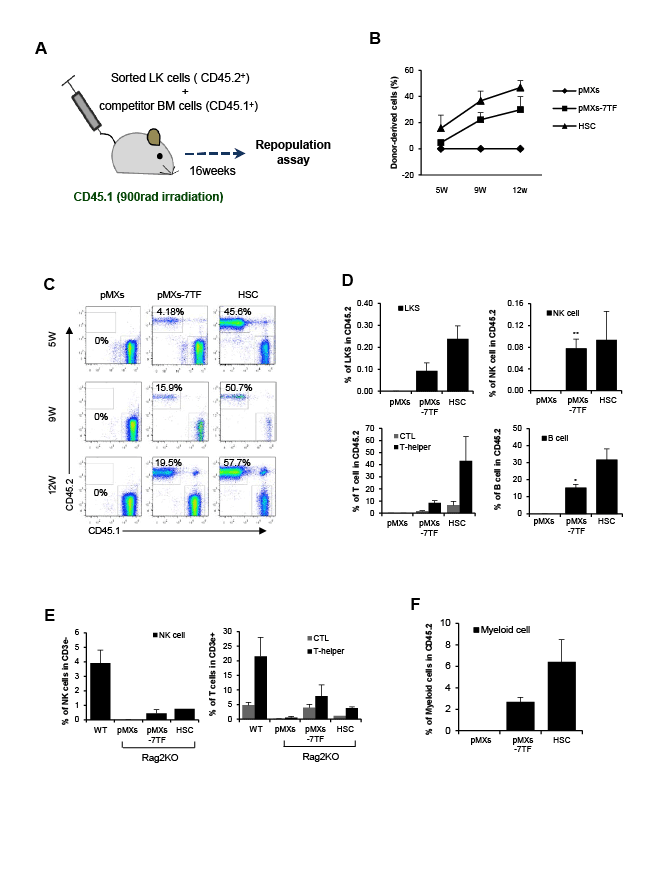
Figure 3: Multipotent capacity of pMXs-7TF MEFs in vivo. (A) The experimental design of the competitive reconstitution assay. Irradiated CD45.1
congenic mice were transplanted with 5 × 105
LK cells from donor-derived BM (CD45.2) or pMXs-7TF MEFs (CD45.2) with competitor cells (CD45.1, 1
× 106
). After 16 weeks, the frequencies of donor-derived LKS, NK, T and B cells were determined via FACS analysis. (B) Peripheral blood was collected
at 5, 9 and 12 weeks after BM transplantation to determine the percentage of donor-derived cells. The error bars represent the s.d. (n=4). (C) Donorderived
cells (CD45.2+) in peripheral blood were analyzed by flow cytometry. (D) Donor-derived (CD45.2+) LKS, NK, B or T cells in the recipient BM
(CD45.1+) at 16 week after transplantation. The error bars represent the s.d. (n=4). *p<0.05 and **p<0.01 compared with pMXs control vector-transduced
MEFs. ; see also Supplementary Figure 7. (E) pMXs-7TF MEFs were transplanted into Rag2 KO mice. After nine weeks, the wild type mice (C57BL/6J,
WT), the Rag2 KO mice (pMXs), and the HSC-transplanted (HSC) or pMXs-7TF MEFs-transplanted (pMXs-7TF MEFs) Rag2 KO mice were euthanized
and analyzed via FACS analysis. The left panel indicates NK cells, and the right panel indicates T cells. ; see also Supplementary Figure 7. The error
bars represent the s.d. (n=3). Representative results from one of three repeated experiments are shown. (F) To confirm the generation of myeloid cell
populations in vivo, we verified the presence of the CD45+GR-1+Mac-1+ cell populations in the donor-derived cells. LK cells (5 × 105
) from iHSCs or BM
cells were transplanted into lethally irradiated congenic mice (CD45.1). After nine weeks, donor-derived myeloid cells (CD45.2+) were analyzed in the
peripheral blood of the recipients (CD45.1+). ; see also Supplementary Figure 7. The error bars represent the s.d. (n=3).
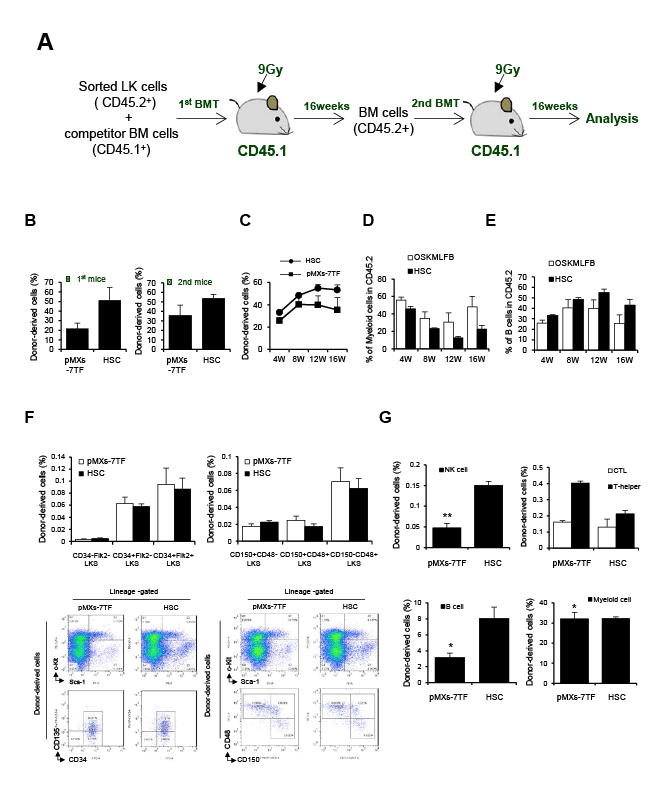
Figure 4: A serial BMT of pMXs-7TF MEFs. (A) The experimental design of serial BMT assay. Irradiated CD45.1 congenic mice were transplanted with
5 × 105
LK cells from donor-derived BM (CD45.2) or pMXs-7TF MEFs(CD45.2) with competitor cells (CD45.1, 1 × 106
). At 16week after the first BMT,
donor-derived BM cells (3×106
) from recipients were injected into a second set of recipient mice (CD 45.1+). (B) PB was collected at 16 week after first
BMT and secondary BMT to determine the percentage of donor-derived cells; see also Supplementary Figure 8. (C) To verify the engraftment of donorderived
cells in the blood, we collected PB at 4, 8, 12 and 16 week after secondary BMT. (D-E) Reconstitution of the indicated populations in PB at 16
week after secondary BMT. Donor chimerism presented as percentage of donor cells. (F-G) Donor-derived (CD45.2+) HSC populations, NK, T, B and
myeloid cells in the recipient BM (CD 45.1+) at 16 week after second BMT assay; see also Supplementary Figure 9. The error bars represent the s.d.
(n=3). *p<0.05 and **p<0.01 compared with HSC
hCD34 expression in mouse LT-HSCs
To further confirm the reprogramming capacity of pMXs-7TF-MEFs,
we used transgenic (tg) mice carrying the hCD34 locus and 12.8 kb of 5’
and 25.6 kb of 3’ flanking regions, which are necessary to direct hCD34
expression exclusively in LT-HSCs [27], such that pMXs-7TF MEFs
regeneration could be monitored based on hCD34 expression. MEFs
obtained from hCD34 tg mice were transduced with a retrovirus carrying
the candidate genes Oct 4, Sox2, Klf4, c-Myc, LMO2, c-Fos and c-Myb.
After obtaining pMXs-7TF MEFs from the hCD34 tg-derived MEFs, the
expression of HSC markers was analyzed. As shown in supplementary
figure 10A, hCD34+ cells were observed in the LT-HSC population. We
then performed qPCR and RT-PCR to verify the expression of hCD34
and the HSC marker genes. c-Kit, Sca-1, CD150 and hCD34 were highly
expressed in the pMXs-7TF-transduced cells, as expected; however,
hCD34 was not expressed in the pMXs-transduced wild type or hCD34
tg MEFs, ES cells, or HSCs (Supplementary figures 10B and 10C). These
results confirmed that pMXs-7TF MEFs are similar to primitive HSCs
with respect to hCD34 gene expression.
Discussion
In this study, we demonstrated that MEFs can be converted to mouse
HPCs via the expression of Lmo2, c-Fos, c-Myb, Oct4, Sox2, Klf4 and
c-Myc. First, the reprogrammed pMXs-7TF MEFs expressed HSC surface
markers, such as c-Kit and Sca-1, and they produced detectable long-term
HSC, short-term HSC and MPP cell populations. Second, the pMXs-7TF
MEFs were maintained in vitro for extended periods. To confirm whether
pMXs-7TF MEFs can undergo long-term expansion, the pMXs-7TF MEFs
were cultured in Myelocult long-term culture medium supplemented with
hematopoietic cytokines. After two months, we observed that the LKS cell
population remained present in the pMXs-7TF-transduced cells. Third,
the gene expression profiles of the pMXs-7TF MEFs were nearly identical
to those of primitive HSCs. HSC surface markers (c-Kit, Sca-1, CD34 and
CD150) and transcription factors (c-Jun, Lmo2, c-Fos, c-Myb, c-Myc and
Trim28) that are highly expressed in HSCs were strongly expressed in
pMXs-7TF-transduced cells, although the pluripotent cell markers SSEA,
Nanog and E-Ras were not detectable in these cells. Fourth, the pMXs-
7TF MEFs displayed the potential to differentiate into multiple lineages in
vivo and in vitro. When the pMXs-7TF MEFs were injected into lethally
irradiated congenic or immunodeficient mice, they exhibited potent
reconstitutive capacities. Moreover, the pMXs-7TF MEFs differentiated
into both the lymphoid and myeloid lineages, which perform distinct
functions. Taken together, these results demonstrated that pMXs-7TF
MEFs can be reprogrammed using specific genes without the need to
generate iPS cells and that these pMXs-7TF MEFs display phenotypic and
functional characteristics of HPCs.
According to the SAGE dataset, the expression of c-Jun, Lmo2,
c-Fos, c-Myb and c-Myc were selectively elevated in HSCs. These TFs
are necessary for the regulation of HSC properties such as self-renewal
and differentiation potential. c-Myc is one of the transcription factors
important for HSC differentiation and survival [39,40] and regulates the
balance between the self-renewal and differentiation of HSCs [41]. Lmo2
and c-Myb are critical for the development of HSCs [42-44], and Lmo2 in
particular is necessary for promoting a hematopoietic fate in embryonic
fibroblasts [23]. c-Fos, a member of the AP-1 transcription factor, in a
complex with c-Jun protein regulates expression of AP-1 binding genes
[45]. AP-1 transcription factor plays multiple roles in development of
hematopoietic precursor cells into mature blood cells including the
granulocyte monocyte and erythroid lineages [46,47]. c-Fos is highly
expressed in HSCs [48] and is important for cell growth and the G0/G2
transition in HSCs. According to recent study, c-Fos promotes endothelial
and hematopoietic gene expression [20]. In the present study, we
identified a combination of HSC-specific transcription factors that induce
the conversion of MEFs to HPCs in the presence of Oct4, Sox2 and Klf4.
In conclusion, the combination of HSC-specific transcription factors
such as Lmo2, c-Fos and c-Myb and pluripotency-related genes such as
Oct 3/4, Sox2, Klf4 and c-Myc can induce the direct conversion of multipotent
iHSCs from MEFs. The present study demonstrates the iHSCs
have the potential to differentiate into immune cells and to repopulate
hematopoiesis in vivo. The success of iHSC generation from the patient’s
own somatic cells will facilitate the treatment of several diseases and
can be used instead of HSCs. This method will accelerate the clinical
application of iHSC generation as a platform technology for regenerating
patient-specific HSCs.
Acknowledgements
The authors are grateful to Daniel G. Tenen (Harvard Medical School)
for providing hCD34 transgenic mouse. This work was supported in part
by research funding from the R&D convergence Program (No. CRC-
15-02-KRIBB) of NST, the GRL project (FGM1401223), the Ministry of
Science, ICT & Future Planning, and the Korean Health Technology R&D
Project (A121934), Ministry of Health and Welfare, and KRIBB Research
Initiative Program, Republic of Korea.
Author Contributions
H.Y.S. designed the experiments and wrote the paper. H.Y.S., H.W.S.,
Y.K.K., W.K. and S.Y. performed the experiments. E.L. generated the
hCD34 transgenic mice. E.L., H.J., S.R.Y., T.D.K., and Y.J.P. provided
critical comments. I.C. supervised the project and wrote the paper.
Conflicts of Interest Disclosure
The authors declare no competing financial interests.
