Introduction
Cardiovascular disease is the leading cause of death in the United States,
and coronary heart disease, which leads to ischemic myocardial injury is
the most common cardiovascular disease with a toll of 370,000 deaths
each year in the US [1]. Cardiomyocyte apoptosis and fibrosis initiated
by an infarct result in continuing adverse remodeling of the myocardium,
which leads to eventual heart failure [2,3]. Current therapies for MI focus
mostly on disease management and prevention of MI recurrence. Hence,
there is a need to develop effective therapeutics that addresses the core
pathophysiology, particularly the ongoing cardiomyocyte death and
fibrosis that occur in the aftermath of an infarct.
The WNT/β-catenin pathway is activated in various cardiac cells in
response to ischemic injury [4,5], and has been studied by many groups
in the context of myocardial infarct repair. In genetic models of WNT
pathway modulation through β-catenin stabilization or depletion in
cardiomyocytes, WNT activation leads to adverse remodeling after
ischemic injury, whereas WNT inhibition improves function [6,7]. Studies
using overexpression or exogenous administration of secreted WNT
inhibitors (secreted Frizzled-Related Protein 1 and 2; sFRP1, and sFRP2
[8,9]) have shown that WNT inhibition stimulates recovery of cardiac
function and reduces scarring after MI, although the cellular mechanisms
proposed by these studies have been different. Distinct and incomplete
blocking of ligand-dependent vs. independent WNT signaling [10], or
in the case of pyrvinium [11,12], incomplete therapy due to toxicity and
effects on other signaling pathways, may account for the differences in
cellular effects of WNT inhibitory therapeutics observed in these studies.
Moreover, there are conflicting reports suggesting that WNT activation
can also enhance post-MI cardiac repair. Paik et al. [13] showed that gainof-function
of a canonical WNT ligand, WNT 10b, in cardiomyocytes can
orchestrate recovery by augmenting neovascularization. Interruption of
WNT 1/β-catenin signals specifically in the epicardium through β-catenin
deletion was reported to impede adaptive pro-fibrotic response [14,15],
whereas post-infarct injection of adenoviral vector with constitutively
active β-catenin reduced infarct size by boosting cardiomyocyte survival,
and granulation tissue formation by myofibroblasts [16]. Some of the
discrepant observations may be explained by ligand [15] and cell-type
dependent [16] effects of WNT signaling on healing. Moreover, the genetic
models that use cell-specific promoters to modulate WNT signaling have
caveats, such as incomplete targeting of the microenvironment, inability
to transiently inhibit the signal, and unintended physiological effects
on the cell types targeted (for example, complete β-catenin mutation in
epicardial cells [14] may lead to deficiencies in the developing heart as
reported in previous studies [17] complicating the investigation of its
effect on the infarcted heart).
We hypothesized that short term WNT inhibition with a pharmacologic
agent that comprehensively targets all WNT ligand-dependent signaling
improves cardiac recovery following MI in a mouse model of permanent
ligation of the left ventricle descending coronary artery by affecting
multiple cardiac cells to mediate repair. The paucity of safe and effective
WNT inhibitors suited for clinical use has delayed efforts to assess
therapeutic WNT pathway inhibition in cardiac injury [18,19]. Recently,
with the new class of WNT ligand secretion inhibitors, that act on the
membrane-bound O-acyltransferase, Porcupine [20], we are able to
investigate the potential of systemic WNT inhibition for post-MI cardiac
injury repair. The enzyme Porcupine palmitoylates WNT ligands—a
modification is essential for the secretion and receptor binding of WNT
proteins [21]. It has been shown that Porcupine activity is specific to
WNT ligands and does not affect other similarly lipid modified signaling
proteins such as Hedgehog [22], hence making it a suitable candidate for
WNT pathway targeting. For our studies, we used GNF-6231, a Porcupine
inhibitor synthesized by the Genomics Institute of the Novartis Research
Foundation. An analog of the compound, LGK974 [21], has already
advanced to clinical studies for WNT-driven cancers [23]. Here we report
that short-term pharmacologic WNT inhibition following experimental
MI augments cardiac repair, characterized by improvement in functional
and remodeling parameters, and reduction in collagenous scar. Our data
suggest that this occurs through effects on multiple facets of cardiac
pathology including, proliferative response in interstitial (possibly,
progenitor) cells in the heart, through reduction in cardiomyocyte
apoptosis, and through mitigation of myofibroblast proliferation and
collagen synthesis.
Materials and Methods
Antibodies
The following antibodies were used: β-catenin (1:200; BD Pharmingen,
610153); β- galactosidase (1:100; AbCam, Ab616); Alpha Smooth Muscle
Actin (α-SMA) (1:1000; Sigma A2547); Ki67 (1:400; AbCam, Ab15580);
phospho-Histone-H3 (1:200; Millipore, 06-570); cTnI (1:1000; AbCam,
Ab6556); Alpha Sarcomeric Actin (1:200; Sigma, A2172); GATA4 (1:200;
Santa Cruz, SC25310); Periostin (1:100; Santa Cruz, SC67233); PCNA
(1:100; Santa Cruz Biotech, SC-56); FSP1 (1:100; Millipore, 07-2274);
Vimentin (1:200; Sigma, V2258); vWF (1:200; Takara, M116).
WNT modulators
The small molecule Porcupine inhibitor GNF-6231 was a generous gift
from the Genomic Institute of the Novartis Research Foundation. Small
molecule WNT inhibitor (CK1α activator) VU-WS113 (C-113) was a
generous gift from Dr. Ethan Lee, Department of Cell and Developmental
Biology, Vanderbilt University [24]. Recombinant mouse WNT3A was
purchased from AbCam (ab81484) or from the Vanderbilt Antibody and
Protein Resource (VAPR). For in vitro studies, GNF-6231 in DMSO as
vehicle was used at a concentration of 100 nM; C-113 (vehicle: DMSO)
was used at 1 µM; and recombinant mouse WNT3A (vehicle: 01% BSA
in PBS) was used at a concentration of 50 ng/mL after testing a range of
concentrations between 25 ng/ml- 100 ng/ml and demonstrating similar
proliferative response (data not shown).
Animals
All procedures were carried out in accordance with Vanderbilt
Institutional Animal Care and Use Committee (IACUC), and NIH
guidelines. C57Bl/6J mice were purchased from the Jackson Laboratory
(Bar Harbor, ME) and maintained by PPY. TOPGAL [5] mice were a
generous gift from Dr. Antonis Hatzopoulos (Department of Cell and
Developmental Biology, Vanderbilt University).
Cell lines
Sca1+CD31-
CD45- cells were isolated as previously described [25].
Briefly, H-2Kb
-tsA58 transgenic mice in C57Bl/6 background expressing
temperature sensitive thermolabile simian virus (SV40) large tumor (T)
antigen under the ubiquitous mosue major histocompatibility complex
(H-2Kb
) promoter—age 6- to 8-weeks—were euthanized using overdose
of Isoflurane followed by cervical dislocation. Hearts from five mice
were dissected to isolate ventricular tissue, which was then minced and
incubated with 10 ml of digestion solution (10 mg/ml collagenase II,
2.5 U/ml dispase II, 1 µg/ml, DNase I, and 2.5 mM CaCl2
) for 20 min at
37°C. The non-myocytes were collected using Percoll gradient. A filtered
myocyte-free single-cell suspension in PBS containing 0.5% BSA and 2
mM EDTA (PBS/BSA/EDTA) was treated with mouse BD Fc Block (clone
2.4G; BD Biosciences, San Jose, CA), and immune cells were magnetically
removed with CD45 microbeads (Miltenyi Biotec Inc., Auburn, CA).
After incubation with phycoerythrin (PE)-conjugated CD31 (clone 390;
eBioscience, San Diego, CA) and fluorescein isothiocyanate (FITC)-
conjugated Sca1 (clone E13-161.7; BD Biosciences) antibodies, CD31
positive cells were removed with anti-PE microbeads (Miltenyi Biotec).
Sca1+CD31- cells were magnetically isolated with anti-FITC microbeads
(Miltenyi Biotec). Isolated conditionally immortalized Sca-1+CD31-
CD45- cells were plated at a density of 104 cell/cm2 and cultured on 1%
gelatin-coated tissue culture dishes in DMEM supplemented with 10%
FBS, 1% Penicillin/Streptomycin, and 2 mM glutamine and 10 ng/ml
IFN-γ under a humidified atmosphere of air/CO2
(19:1) at 33°C. Six days
before experiments, cells were replated and cultured in the absence of
IFN-γ at 37°C.
HL1 cell line, derived from mouse atrial cardiomyocytes was a
kind gift from Dr. William C. Claycomb (Louisiana State University
Medical Center, New Orleans, LA). These cells were cultured on gelatin/
fibronectin (25 µg fibronectin in 2 ml of 0.02% gelatin in water)- coated
plates (fibronectin and gelatin from Sigma-Aldrich). The HL1 cell line was
maintained at 37°C in Claycomb medium (SAFC Biosciences, Lenexa,
KS) supplemented with 10% fetal bovine serum (SAFC Biosciences),
100 µM norepinephrine (Sigma) in 30 mM ascorbic acid (Sigma), 2 mM
L-Glutamine (Sigma), penicillin, and streptomycin (Life Technologies,
Grand Island, NY).
iCell Plus Cardiomyocytes, which are primarily ventricular
cardiomyocytes derived from human induced pluripotent stem (iPS) cells,
were purchased from Cellular Dynamics International and maintained
in 0.1% gelatin coated plastic plates in manufacturer’s proprietary
maintenance medium.
Primary mouse cardiac fibroblasts were isolated from the hearts
of C57Bl/6 mice that were at least 12 weeks old following a previously
described protocol [26]. Briefly, mice were euthanized by overdose of
isoflurane followed by cervical dislocation. Heart tissue was minced and
placed into Kreba-Henseleit (Sigma; K3753a) buffer with 2.9 mM CaCl2
and 24 mM NaHCO3 containing a cocktail of 0.25 mg/mL Liberase
Blendzyme 3 (Roche Applied Science), 20 U/mL DNase I (Sigma Aldrich),
10 mmol/L HEPES (Invitrogen) and 0.1% sodium azide in HBSS, and
shaken at 37°C for 20 min. Cells collected after digestion were passed
through 40 µm nylon mesh and centrifuged (15 min, 200 g, 4°C). Finally,
cells were reconstituted with DMEM-F12 medium containing 10% FBS
and 1% Penicillin/Streptomycin and seeded onto plastic plates (Corning)
for separation of fibroblasts by selective adhesion for 4 hours at 37°C. Cells
were maintained in culture medium composed of high glucose (4.5 g/L)
DMEM with 10% FBS and 1% Penicillin/Streptomycin under a humidified
atmosphere of air/CO2 (19:1) at 37°C.
Proliferation assay
Cell proliferation was assessed by 5-bromo-2’-deoxyuridine (BrdU) cell
proliferation assay (Calbiochem, Gibbstown, NJ). Briefly, CPCs, HL-1 cells,
or primary fibroblasts were seeded on 96-well plates (gelatin/fibronectincoated
for HL-1 cells), with recombinant WNT3A where indicated.
Following attachment onto plates, WNT modulators were added and
cultured for 24 hours. BrdU incorporation was assessed by enzyme-linked
immunosorbent assay (ELISA) and read at a dual wavelength of 450/595
nm using the SOFTMax Version 2.35 software (Molecular Devices, LLC,
Sunnyvale, CA) following manufacturer’s recommendations.
Cell viability assay
HL-1 cardiomyocytes were serum starved overnight in 2% FBS, and
seeded onto (gelatin/fibronectin-coated for HL-1) 96-well plate at a
density of 104 cells/well. Following attachment of cells over 24 hours, WNT
modulators (50 ng/mL recombinant mouse WNT3A with or without 1
µM C-113) were added and the cells incubated for 48 hours. Cell viability
was assessed by incubating with 0.5 mg/mL of 3-[4,5-dimethylthiazol-2-
yl] 2,5,-diphenyltetrazolium bromide (MTT) (Sigma) in PBS for 4 hours
at 37°C, and MTT reduction into formazan by viable cells was measured.
Formazan crystals were dissolved in 75 µL/well DMSO 10 minutes at
37°C. Photometric measurement was carried out at 540 nm. Percentage
viability was calculated by as follows: %Cell survival=(ODControl-ODSample)
× 100%/ODControl. Untreated cells were used as control. Results represent
three independent experiments, each performed in triplicate.
Surgical LAD ligation (myocardial infarction) model, drug
treatment, echocardiography and infarct size calculations
Male C57Bl6J mice (at least 3 months of age) were anesthetized under
sodium pentothal (50 mg/kg) and endotracheal intubation was performed
under direct laryngoscopy. Mice were ventilated with a small animal
respirator (tidal volume=1.0 ml, rate=110 breaths/min). With the use
of a surgical microscope, a left thoracotomy was performed. The fourth
intercostal space was entered using scissors and blunt dissection. A 7-0 silk
suture was placed through the myocardium into anterolateral LV wall and
the left anterior descending artery was ligated. The heart was monitored
with continuous EKG throughout the procedure to ensure successful
infarction. The chest was closed in layers with 6-0 silk (6-0 nylon to close
the skin) and the animal was gradually weaned from the respirator to avoid
complicating pneumothorax. Intraperitoneal administration of 0.1 mg/kg
buprenorphine immediately following surgery and every 8-12 hours for
72 hours post-surgery was used as analgesic. Animals were monitored
closely for signs of distress and weight loss throughout the study period.
Severe respiratory distress, lack of ambulation, or severe weight loss (>20-
30% of body weight) was considered reasons for euthanizing the animal.
Following study completion, or upon distress, animals were euthanized by
overdose of isoflurane followed by cervical dislocation.
Starting from within 6 hours after surgery through day 6 post-infarct,
mice were treated every 24 hours with intravenous (tail-vein) injection
of 5 mg/kg (100 µL volume) GNF-6231 or vehicle (3% D-α-tocopheryl
polyethylene glycol succinate or Vitamin E in 20% Polyethylene glycol).
For cardiac recovery study, cardiac dimensions were obtained from
2-D guided M-mode images (100 frames/sec) and were read blinded
using short axis and a parasternal long-axis views. All measurements were
done on unsedated mice at day 7 and day 30 post-MI. Measurements were
averaged over 3 consecutive beats from the LV posterior wall (LVPW)
the interventricular septum (IVS) and LV internal diameter (LVID).
After day 30 echocardiography, hearts were excised, immersion-fixed
in and paraffin-embedded to obtain serial sections in order to measure
the infarct size (% area of tissue stained blue for collagen in H&E stained
sections) in a blinded manner.
Separately, for longitudinal studies, hearts were excised at day 3,
7, and 15 days post MI, and processed to obtain paraffin sections for
immunostaining studies.
Histology and morphometry
Hearts were fixed in 10% buffered formalin for 24 hours, embedded
in paraffin and sectioned into 5 microns-thick transverse sections.
H&E and Masson’s trichrome staining was performed by the Vanderbilt
Translational Pathology Shared Resource. Olympus DP71 microscope
camera (Olympus America, Center Valley, PA) was used for imaging H&E
and Masson’s trichrome stained sections.
For immunofluorescence staining, slides were deparaffinized and
hydrated through xylene and ethanol steps. Heat-mediated antigen
retrieval was performed by boiling in citrate buffer (pH 6). Cells
seeded onto coverslips were fixed for 1 hour at room temperature in 1%
paraformaldehyde, permeabilized with 0.1% Triton-X in 0.1% sodium
citrate for 2 min on ice, and washed several times with PBS. Following
blocking with 10% goat serum in 1% BSA solution for 1 hour at room
temperature, sections were incubated with primary antibody at 4°C
overnight, and Alexa Fluor 488 or Cy3 conjugated secondary antibodies
at room temperature for 1 hour. The slides were then counterstained with
Hoechst 33342 (H21492 Invitrogen, Carlsbad, CA) and mounted with
Slowfade Gold (S36936 Life Technologies, Grand Island, NY). For TUNEL
staining, a 1:10 mix of enzyme:label diluted 5 fold with TUNEL dilution
buffer (In Situ Cell Death Detection Kit TMR Red, Roche 12 156 792 910)
was added to samples along with the secondary antibody and incubated
for 60 minutes at 37°C. Samples were then counter stained with Hoeschst
and mounted. Images were taken at 10x, 20x or 40x magnification using
Axio Imager2 microscope (Carl Zeiss, Thornwood, NY) and CoolSNAP
HQ CCD camera (Photometrics, AZ), and quantified using ImageJ. For
confocal microscopy, LSM510 (Zeiss) microscope was used to capture 1
µm optical slices. All images are presented with scale bars that equal 50
µm.
PK/PD study
Single dose PK/PD profile of GNF-6231 was investigated in C57Bl/6
mice following a 5 mg/kg intravenous bolus injection following guidance
from manufacturer. The drug was dissolved in 20% Poly Ethylene Glycol
(PEG) 300 with 3% D-α-Tocopherol polyethylene glycol 100 succinate
(ETPGS) in H2
O. Briefly, GNF-6231 was homogenized using QIAGEN
bead homogenizer for at least 10 minutes in the presence of 25% of the final
volume of the vehicle to be used to form slurry. Another 25% of vehicle
was added and bath sonicated on high for 10 minutes. The remaining
volume of vehicle was added and vortexed. An additional 10 minute bath
sonication was performed. Further, probe sonication was performed at
50% amplitude for 3X 30 seconds on ice bath. The reconstituted drug was
filtered with a 0.45 µm syringe filter. Vehicle alone underwent similar
preparation in the absence of GNF-6231. At specific time points following
intravenous administration, blood was collected from the saphenous
vein. Plasma concentrations of GNF-6231 were quantified by LC/MC/MS
analysis. Briefly, aliquots of plasma samples were added to the internal
standard and acetonitrile/methanol (3/1), samples were vortexed and
centrifuged at 4,000 rpm for 5 minutes at 4°C to precipitate the plasma
proteins. Supernatant was transferred to a clean 96-well plate, and diluted
with distilled water. The extracted samples were injected (10 µL) onto a
Zorbax SB-C8 analytical column (2.1 × 30 mm, 3.5 µm).
Agilent Technologies Inc., Palo Alto, CA, USA). Mobile phases
consisted of 0.05% formic acid in water (solvent A) and 0.05% formic acid
in acetonitrile (solvent B), and a gradient elution method at a flow rate of
700 µL/min was used for compound elution and separation. Mass spectral
analyses were carried out using atmospheric pressure chemical ionization
(APCI) in the positive ion mode, with multiple reaction monitoring
(MRM) of GNF-6231 (449.2>221.0). The lower limit of quantitation
(LLOQ) in plasma was 1.0 ng/mL. Pharmacokinetic parameters were
calculated by non-compartmental regression analysis using an in house
fitting program.
PD study was performed as described previously [21]. Briefly, livers
were collected from mice at specific time points after GNF-6231 or vehicle
injection following euthanasia with isoflurane overdose and cervical
dislocation. Total RNA was isolated using the Qiagen RNeasy kit; TaqMan
analyses were performed using Axin2 and Gapdh probes (Applied
Biosystems) according to the manufacturer’s instructions. mRNA
expression levels for the target gene, Axin2, was normalized to Gapdh
mRNA levels and data were analyzed using SDS 2.0 software (Applied
Biosystems) to calculate relative RNA quantities.
RNA isolation and qRT-PCR
RNA was isolated from cells were using Trizol Reagent (Invitrogen,
15596026) following manufacturer’s protocol. First strand DNA synthesis
was performed with 1 µg RNA using iScript cDNA synthesis kit (Bio-Rad
170-8890). Quantitative real-time PCR was performed in triplicate for
each sample with iCycler (BioRad) and fluorescence detection (SsoFast
EvaGreen; 172-5200; BioRad). Each reaction was normalized against 18S.
Primer sequences are provided in supplementary Table S1.
Statistical analysis
The statistical significance between experimental and control groups
were determined by One-way ANOVA with Bonferroni correction
for multiple comparisons when multiple groups were compared. The
D’Augustino and Pearson omnibus or the Shapiro-Wilk tests were used to
determine whether the data sets were normally distributed. For data sets
that were not normally distributed or had N<7, the Kruskal-Wallis H-test
was used instead of One-way ANOVA. For comparison between two
groups of data, unpaired t-test was used for normally distributed datasets,
and Mann-Whitney test was used for data that were not normally
distributed. GraphPad Prism (San Diego, CA) software was used for all
statistical analyses. P<0.05 was considered statistically significant in twotailed
hypothesis tests.
Results
Porcupine inhibitor GNF-6231 downregulates WNT target
gene expression, is bioavailable in vivo, and is well-tolerated by
WNT-dependent tissues
In order to investigate the physiological and cellular effects of WNT
inhibition on post-MI cardiac regeneration, we utilized GNF-6231, a
small molecule inhibitor of the acyltransferase, Porcupine. Porcupine is
the enzyme responsible for the post-translational palmitoylation of WNT
proteins that is required for both WNT secretion as well as binding of
WNTs to their receptors [21] (Figure 1A). An analog of the compound
is currently in Phase I clinical trials as WNT inhibitory therapeutics
for cancer [23]. GNF-6231 inhibits Porcupine enzymatic activity with
a cellular IC50 of 0.8 nM, and does not show cytotoxicity up to 20 µM
[27]. In Wnt3a overexpressing cardiac cells, it potently inhibited WNT
pathway activation as indicated by Axin2 mRNA levels (Figure 1B). In
vivo, the pharmacokinetic (PK) and pharmacodynamic (PD) relationship
of GNF- 6231 was investigated following a single 5 mg/kg intravenous
administration of GNF-6231 or vehicle to C57BL/6J mice. GNF-6231
showed high plasma levels, and free plasma concentrations (mouse
plasma protein binding of GNF-6231 is 88%) above its in vitro Porcupine
IC50 for at least 12 h. Plasma half-life of GNF-6231 was estimated to be
2.3 hours (Figure 1C). The expression of the WNT target gene Axin2 was
measured in liver tissues. Although reduction in Axin2 gene expression
in the liver by GNF-6231 treatment started by 3 hours after a single
intravenous injection, a statistically significant reduction occurred at 7
hours post-treatment. At 24 hours, there was a 37% reduction in Axin2
expression compared to vehicle, indicating successful WNT inhibition
at that time point (Figure 1D). Since GNF-6231 inhibits the enzymatic
activity of Porcupine, impeding WNT secretion, an expected time delay
was observed between peak GNF-6231 plasma concentration (5 min) and
the PD response as measured by Axin2 inhibition (Figure 1D).
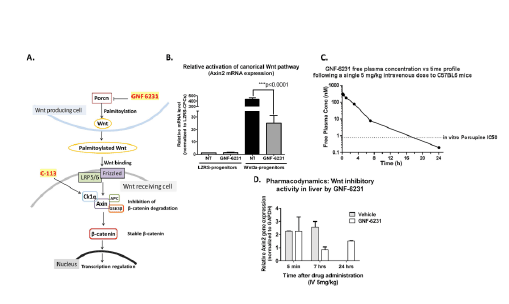
Figure 1: GNF-6231 inhibits canonical WNT pathway activity in vitro. (A) Schematic of the WNT pathway and point of action of WNT inhibitors,
GNF-6231 and C-113. (B) Fold change in Axin2 gene expression in WNT3a overexpressing cardiac cells showing GNF-6231 treatment reduced
WNT target gene expression (N=3 replicates from independent experiments; ***p ≤ 0.0001; Repeated measures ANOVA with Bonferroni correction
for multiple comparisons). (C) IV free plasma level of GNF-6231 after a single intravenous injection of 5 mg/kg. The plasma half-life of the drug was
approximately 2.3 hours; GNF-6231 free plasma concentrations were above the in vitro Porcupine IC50 for >12 h. (D) qRT-PCR showed inhibition of
Axin2 gene expression in liver at different time points following a single 5 mg/kg intravenous treatment with GNF-6231 (N=2 mice per timepoint). Bars
represent Mean ± SD.
Since the canonical WNT pathway is constitutively active in certain
tissues such as colon and skin, we assessed the potential toxicity of
inhibiting WNT signaling in these tissues. With 6 daily consecutive
treatments of 5 mg/kg GNF-6231 intravenously (dosage and regimen
used in our studies; described in Figure 2A), there was no effect on the
histology of the colon (Supplementary Figure 1A), or β-catenin expression
and localization as detected by immunostaining (Supplementary Figure
1B), signifying no GI tract toxicity of the drug. Likewise, no effect on skin
histology (Supplementary Figure 1C) was observed in GNF-6231 treated
animals compared to vehicle- treated controls.
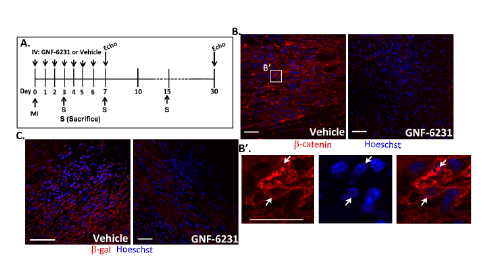
Figure 2: Porcupine inhibitor treatment inhibits WNT pathway activity in the infarcted heart. (A) Schematic summarizing animal study timelines.
Mice were treated with daily intravenous injection of 5 mg/kg drug or vehicle following MI and continued through day 6. For cardiac recovery studies,
mice underwent echocardiography at days 7 and 30. For histology, a separate cohort of mice was sacrificed on days 3, 7 and 15. (B) β-catenin
immunostaining of peri- infarct region of ventricles at day 7 showed reduction in β-catenin levels with GNF-6231 treatment. (B’) High magnification
image of vehicle-treated tissue showed nuclear localization of β-catenin signifying WNT pathway activation. (C) β-galactosidase immunostaining in
ventricle sections from WNT reporter, TOPGAL mice demonstrated inhibition in WNT activity at day 7 post-infarct with GNF-6231 treatment. Scale bars
equal 50 µm. Images are representative of sections from N ≥ 3 mice; at least 4 areas were imaged from each mouse.
To confirm our findings with GNF-6231 in vitro, we used a small
molecule Casein Kinase1-alpha (CK1α) activator VU-WS113 [24],
referred to in this paper as C-113. It targets the β-catenin degradation
complex [24,28], and hence inhibits the WNT pathway by a mechanism
of action that is distinct from GNF-6231 (Figure 1A). Quantitative realtime
PCR for WNT target gene, Axin2 showed that C-113 inhibited WNT
pathway activation induced by treatment with recombinant WNT3A
(Supplementary Figure 2). Since C-113 targets the WNT pathway
downstream of WNT ligand secretion, it allowed us to investigate the
effect of WNT inhibition without the need to overexpress Wnt3a.
Treatment with GNF-6231 inhibits post-MI WNT/β-catenin
pathway activation in the infarcted heart and improves post-MI
recovery/repair
Previous studies have shown that the canonical WNT pathway
is activated in the infarcted heart starting around 72 hours postexperimental
MI [4,5]. WNT pathway activation is reported to peak
between 7 to 14 days post-injury, after which it begins to recede to baseline
levels [5]. To avert this early, transient post-injury WNT activation, we
treated mice (C57Bl6 and WNT reporter, TOPGAL mice that express
β-galactosidase driven by TCF/LEF promoter [5]; age ≥ 12 weeks) with
intravenous injection of 5 mg/kg GNF-6231 or vehicle (3% Vitamin E
and 20% PEG 300) every 24 hours through day 6 after injury (Figure 2A).
The dose and treatment regimen were determined based on our PD/PK
studies and the timeline of WNT activation post-infarct described by
previous studies. Immunostaining for β-catenin (in C57Bl/6J), and for
β-galactosidase (in TOPGAL mice) showed that GNF-6231 treatment
reduced nuclear and cytoplasmic β-catenin levels (Figures 2B and 2B’),
and total β-galactosidase protein levels (Figure 2C) in the peri-infarct
region compared to vehicle treatment. Furthermore, treatment with GNF-
6231 reduced nuclear β-catenin activation in cardiomyocytes themselves
(Supplementary Figure 3).
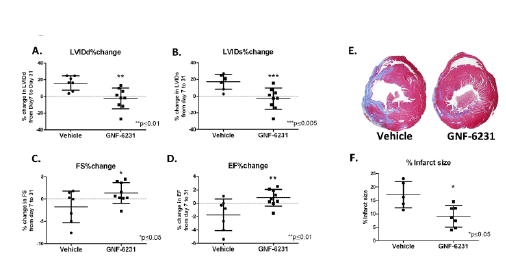
Figure 3: Porcupine inhibition improves cardiac function and reduces adverse remodeling after MI. Left ventricular remodeling was measured as
% change in (A) LVIDd and (B) LVIDs. LV function was measured as % change in (C) FS and (D) EF. Data showed no increase in left ventricular diameter
(A and B), and improved cardiac function (C and D) with GNF-6231 treatment compared to vehicle. (E) Masson’s trichrome stained representative
sections of the left ventricle at day 30 depicted more collagen stained (blue) area in vehicle-treated LV compared to GNF-6231-treated. (F) Quantification
of infarct size. Each data point on graphs represents individual mouse; *p ≤ 0.05, **p ≤ 0.01 or ***p ≤ 0.005; unpaired t-test.
In order to determine the physiological effect of temporary postMI
WNT inhibition, cardiac function and remodeling were assessed at
day 7 and day 30 post-MI using echocardiography (Table 1). Heart rate
was also measured by echocardiography immediately after MI (just
after administration of vehicle or drug) and after 7 days and was not
statistically different between drug and vehicle cohorts (data not shown).
Left ventricular internal dimensions at diastole and systole (LVIDd and
LVIDs respectively) were used as measures of cardiac remodeling. Day 7
measurements enable comparison between cohorts prior to any significant
repair has ensued. The absence of any statistical differences among cohorts
at day 7 support that the degree of MIs were not statistically different
among experimental cohorts (Table 1). At day 30 post-MI, the GNF-6231
treated hearts had lower LVIDd and LVIDs compared to vehicle-treated
mice (LVIDd: 3.83 ± 0.45 mm vs. 4.32 ± 0.68 mm, p=0.0377; LVIDs: 2.35
± 0.38 mm vs. 2.84 ± 0.64 mm, p=0.0446; Table 1). To further control for
variations in infarct size between mice within the experimental groups, we
calculated percent change in each of the parameters from day 7 to day 30
for each individual mouse. The percent change in both of the parameters
of ventricular remodeling (∆LVIDd% and ∆LVIDs %) were significantly
lower in GNF-6231 treated vs. vehicle-treated hearts (∆LVIDd%: -2.287
± 12.36 vs. 16.109 ± 8.53, p=0.0024; ∆LVIDs%: -3.011 ± 12.65 vs. 17.198
± 8.91, p=0.0015; Figures 3A and 3B, Table 1), indicating that WNT
inhibition prevented adverse ventricular remodeling. Ejection Fraction
(EF) and Fractional Shortening (FS) are measures of cardiac function. At
day 30 post-MI, GNF-6231 treated mice had higher EF (0.75 ± 0.05 vs.
0.71 ± 0.06, p=0.0421), and higher FS (38.71 ± 4.13% vs. 34.89 ± 4.86%,
p=0.0325; Table 1) compared to vehicle-treated controls. The percent
change from day 7 to 30 in both parameters of cardiac function (∆EF%
and ∆FS%) were on average, significantly higher for each mouse in GNF-
6231 treated group compared to vehicle-treated group (∆EF%: 0.83 ±
1.25 in GNF-6231 treated vs. -1.723 ± 2.36 in vehicle-treated, p=0.0138;
and ∆FS%: 1.4 ± 2.312 in GNF-6231 treated vs. -1.713 ± 3.59 in vehicle
treated, p=0.0265; Figures 3C and 3D, Table 1), suggesting that WNT
inhibition prevented worsening of cardiac function in the injured heart.
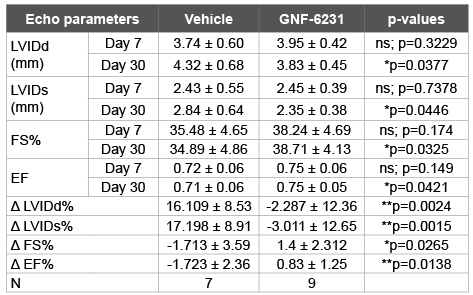
Table 1: GNF-6231 treatment improves cardiac recovery post-MI The top
eight rows represent mean ± SD values for each parameter and treatment
at day 7 and 30. The mean ± SD percent difference between day 7 and
day 30 for each mouse (Δ) are listed in the bottom four rows. Statistical
difference between parameters in each two columns was determined by
unpaired t-test.
The percent infarct area, as determined by blinded histomorphometry
of Masson’s trichrome stained left-ventricular sections (Figures 3E and
3F) by a pathologist at day 30, was significantly lower in GNF-6231 treated
hearts compared to vehicle control (9.07 ± 3.99% in GNF-6231 treated vs.
17.18 ± 4.97% in vehicle-treated; p=0.0152), indicative of a reduction in
myocardial scarring with WNT inhibition. Hence, GNF-6231 augmented
overall cardiac repair and recovery following LV infarct.
WNT inhibition causes proliferation of interstitial cells in the
infarcted heart.
Based on previous studies reporting an anti-proliferative effect of
the WNT pathway in models of skeletal muscle [29] and cardiac injury
[30], we asked whether the reparative effects of GNF-6231 treatment was
mediated in part by proliferation of specific cardiac cells. Immunostaining
for proliferation markers Ki67 and phospho-Histone-H3 (pHisH3)
showed that in the peri-infarct region (defined in Figure 4A), there was
a remarkable increase in pHisH3+ cells at day 3 in both GNF-6231 and
vehicle-treated hearts (Figure 4D). At day 7, the proliferative response was
significantly reduced, but there were 2.3 fold more pHisH3+ cells in the
peri-infarct region of GNF-6231 treated hearts, although the difference
was not statistically significant (Figure 4D). By day 15, the proliferative
response had largely subsided in both treatment groups. In the distal
myocardium however, GNF-6231 treated hearts had significantly more
pHisH3+ cells compared to vehicle-treated at both day 3 and day 7 (5.67-
fold higher; ***p ≤ 0.001, and 2.65-fold higher *p ≤ 0.05 than control
respectively; Figures 4B-4D).
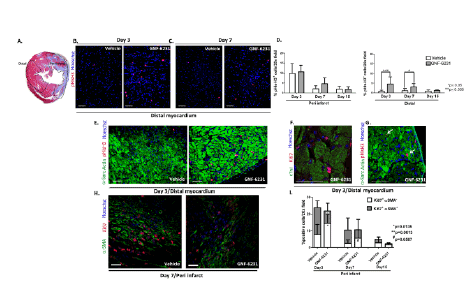
Figure 4: WNT inhibition promotes proliferation of interstitial αSMA negative cells in the infarcted heart. (A) H&E stained cross-section of the
heart demarcating peri-infarct and distal regions of the left ventricle as defined in the study. Representative pHisH3 stained sections of the ventricles
at (B) day 3 and (C) day 7 showing more proliferative cells in the distal myocardium of GNF-6231 treated hearts. (D) Quantification of percent pHisH3+
cells. Bars represent mean ± SD; N ≥ 4 images of sections from N ≥ 3 mice per group were imaged; *p ≤ 0.05, ***p ≤ 0.005; One-Way ANOVA with
Bonferroni Correction for multiple comparisons. Representative sections of the distal myocardium at day 3 post-MI (E) co-stained with αSarcomeric
Actin and pHisH3, and (F) high magnification confocal microscopy image of ventricle co-stained with cTnI and Ki67, demonstrating that the majority
of proliferative cells in the GNF-6231-treated tissue localized to the interstitium of myofibers. (G) αSarcomeric Actin/pHisH3 co-stained LV depicting
the rare proliferating cardiomyocytes (white arrows). (H) Proliferating myofibroblasts were identified by αSMA/Ki67 co-staining as depicted in the
representative section from the peri-infarct region at day 7. (I) Quantification of αSMA/Ki67 co-stained cells revealed that the percentage of proliferating
myofibroblasts (grey shaded portion of the bars) was significantly lower in GNF-6231-treated peri-infarct tissue than control at day 3 (**p=0.0013)
and lower (#
p=0.0587) at day 7. In contrast, the percentage of proliferating non-myofibroblasts (αSMA- cells; lower white portion of the graphs) was
significantly higher (*p=0.0135) in GNF-6231 treated ventricles compared to control at day 7. Bars represent mean ± SD. N ≥ 12: at least 3 separate
sections from at least 3 mice per group were imaged. P-values for individual comparisons between each two groups of data were calculated using
Mann-Whitney test. Scale bars equal 50 µm.
Co-immunostaining for cardiomyocyte marker Alpha Sarcomeric
Actin (αSA) with pHisH3 indicated that most of the cells that were
proliferating in the distal myocardium (higher in proportion in GNF-
6231 treated ventricles) were interstitial cells in both treatment and
control groups (Figure 4E). This was verified by co-staining for cardiac
Troponin I (cTnI) and Ki67; a representative high magnification confocal
microscopy image is shown in Figure 4F. We observed rare cardiomyocytes
with nuclear pHisH3 staining in both GNF-6231 treated and control
animals (a representative example is shown in Figure 4G), but it wasn’t
clear whether these were only undergoing karyokinesis or were truly
dividing cardiomyocytes. Similarly, in vitro BrdU (Bromodeoxyuridine)
incorporation assay with HL-1 cardiomyocyte cell line showed that
recombinant WNT3A and/or WNT inhibitor, C-113 had no effect on
cardiomyocyte proliferation (Supplementary Figure 4C). Since the
proliferating cardiomyocytes were so rare, we focused our investigations
on identifying the interstitial proliferative cells.
WNT inhibition selectively reduces proliferation of
myofibroblasts in the distal myocardium
In the infarcted heart, αSMA-positive myofibroblasts are the major
matrix producing cells responsible for granulation tissue formation
and fibrosis [16]. Hence, we performed co-immunostaining for αSMA
and Ki67 (Figure 4H). Not unexpectedly, proliferating myofibroblasts
(αSMA and Ki67 double positive cells) were present in both the GNF-6231 and vehicle-treated tissue, since some level of pro-fibrotic signaling
is necessary to initiate granulation tissue formation and prevent infarct
rupture [14]. Interestingly, in the peri- infarct region, the proportion of
proliferating myofibroblasts (Ki67+αSMA+ cells; upper dark portions of
the bar in Figure 4I) was significantly lower in the GNF-6231 treated
hearts (2.21 fold lower in GNF-6231 treated; **p=0.0013) at day 3
(Figure 4I). However, the proportion of αSMA negative proliferating
cells (Ki67+αSMA-
, lower white portions of the bar in Figure 4I) in the
peri-infarct region was higher in the GNF-6231 treated hearts than in
vehicle-treated hearts at day 3 and day 7 post infarct (2.7-fold; p=*0.0135
and 2.2 fold; #p=0.0587 respectively; Figure 4I). Likewise, in the distal
myocardium, the proportion of αSMA negative proliferating cells
(Ki67+αSMA-cells)
was 3.3-fold higher in the GNF-6231 treated hearts than
the vehicle-treated hearts at day 3 (*p=0.012) post-infarct (Supplementary
Figure 5A). Meanwhile, co- immunostaining of proliferating cell nuclear
antigen (PCNA) or Ki67 with markers of other fibroblast cell populations,
fibroblast-specific protein-1 (FSP-1; Supplementary Figures 5B and 5C),
Periostin (Supplementary Figure 5D) and Vimentin [31] (Supplementary
Figure 5E) showed no effect of GNF-6231 treatment on proliferation
of these cells. Based on these observations, we posit that GNF-6231
treatment selectively reduced myofibroblast proliferation in the infarcted
hearts, while promoting proliferation of other interstitial cells that did not
include FSP-1+, Periostin+ or Vimentin+ fibroblasts.
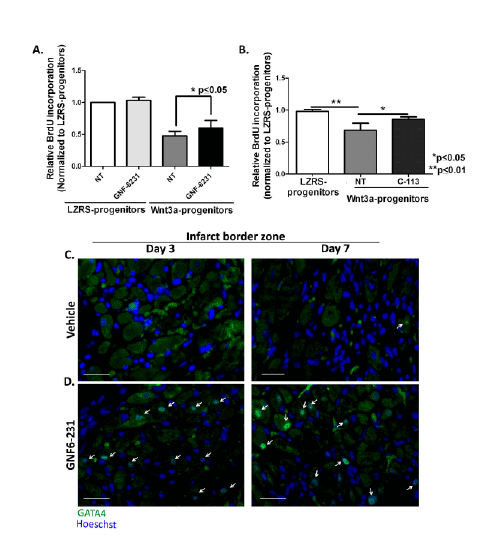
Figure 5: WNT inhibition increases proliferation of progenitor cells that may contribute to myogenesis. (A and B) Relative BrdU incorporation
by Sca1+ progenitor cells stably expressing LZRS (empty vector) or Wnt3a-LZRS revealed that proliferation was reduced by Wnt3a overexpression and
this effect was reversed by (A) GNF-6231 treatment and (B) C-113 treatment.
Data are presented as Mean ± SD. (A) N=5 and (B) N=3 replicates from independent experiments; *p ≤ 0.05 and **p ≤ 0.01; Kruskal-Wallis test with
Dunns correction for multiple comparisons. (C and D) Representative GATA4 immunostained sections of infarct border zone at day 3 (left panels) and
day 7 (right panels) post-MI of (C) vehicle-treated hearts and (D) GNF- 6231 treated hearts. White arrows point to GATA4 stained nuclei. Scale bars
equal 50 µm; the images are representative of at least 4 sections each from N ≥ 3 mice per group.
WNT inhibition increases proliferation of cardiac-derived
Sca1+ progenitor cells
We next sought to determine the identity of the proliferating interstitial
cells that were higher in number in the GNF-6231 treated hearts. To
assess whether these proliferative interstitial cells were endothelial cells
lining the coronary vasculature, co-immunostaining for von Willebrand
factor (vWF) and PCNA was performed. Although a small percentage
of proliferating endothelial cells were observed in both GNF-6231 and
vehicle treated hearts, there was no significant difference in the double
positive cells between the drug and vehicle treated groups (Supplementary
Figures 6A and 6B).
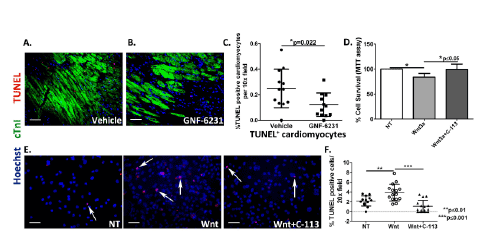
Figure 6: WNT inhibition reduces cardiomyocyte cell death. (A and B) Representative sections co-stained with cTnI and TUNEL of infarct border
zone of (A) vehicle-treated hearts, and (B) GNF-6231 treated hearts. (C) Quantification of percent TUNEL positive cardiomyocytes revealed significantly
fewer apoptotic cardiomyocytes in GNF-6231 treated hearts. N=5 mice per group, at least 4 sections imaged per mouse, *p=0.022, unpaired t-test.
(D) Percent cell survival of HL-1 rat cardiomyocytes as measured by metabolic uptake (MTT) assay showed significant reduction in survival with
recombinant WNT3A treatment, which was rescued by addition of C-113. Bars represent mean ± SD; N=3 replicates from independent experiments;
*P ≤ 0.05; Kruskal- Wallis test with Dunns correction for multiple comparisons. (E) Representative images of human iPSC-derived iCell cardiomyocytes
treated with recombinant WNT3A or WNT3A and C-113 in presence of 250 µM H2
O2
showing that under stress, treatment with recombinant WNT3A
increased cell death, which was rescued by WNT inhibition with C-113. (F) Quantification of %TUNEL positive iCell cardiomyocytes per 20x field. N
≥ 12 per group, at least 4 areas imaged in each replicate from 3 independently run experiments; **p ≤ 0.01 and ***p ≤ 0.001; One-Way ANOVA with
Bonferroni correction for multiple comparisons. Scale bars in A-B and E represent 50 µm.
We and others have shown that WNT inhibition causes proliferation
of adult stem/progenitor cells—bone-marrow-derived MSCs [32] and
cardiac tissue resident-side population progenitors [30]. Additionally,
some of the proliferating interstitial cells in the immunostained sections
of distal myocardium were spherical with large nuclei, and were localized
in what appeared similar to ‘stem cell niche’ for tissue-resident progenitor
cells described in the literature [33] (Figure 4F). These proliferative
αSMA negative interstitial cells were significantly higher in proportion at
the distal myocardium in GNF-6231 treated tissue compared to vehicle
treated myocardium as discussed in the previous section (Supplementary
Figure 5A). We hypothesized that post-injury treatment with GNF-6231
induced proliferation of a cardiac progenitor population.
We assessed the effect of WNT inhibition on Sca1+CD31-
CD45-
CD117-
cells isolated from murine heart homogenates by Fluorescent Activated Cell
Sorting (FACS; Supplementary Figure 7A). Mice expressing thermolabile
simian virus SV40 T antigen (H-2Kb-tsA58 transgenic or Immorto mice)
were used for this purpose [25]. The conditionally immortalized cells
isolated from these mice, under non-permissive conditions do not express
the T-antigen and behave as primary cells (25). These cells were tested for
multipotency based on their ability to differentiate into all three major
cell types in the heart: cardiomyocytes, fibroblasts and endothelial cells
(Supplementary Figures 7B and 7C).
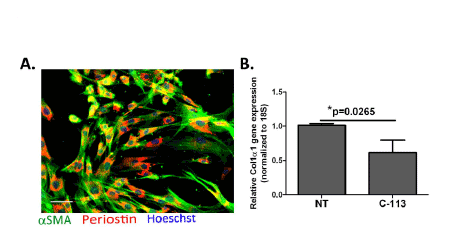
Figure 7: WNT inhibition reduces collagen synthesis activity in vitro. (A) Primary myofibroblasts in culture confirmed by αSMA (green) and
Periostin (red) staining; scale bar equals 50 µm. (B) Relative Col1α1 gene expression in primary cardiac myofibroblasts with and without WNT inhibitor
treatement revealed significant reduction in Col1α1 gene expression in response to WNT inhibitor treatment. Bars represent mean ± SD. N=4 replicates
from independent experiments; *p=0.0265; Mann-Whitney test.
Sca1+ cells stably overexpressing WNT3a were generated in order to
assess the proliferative response to WNT activation and subsequent
inhibition with GNF-6231 or C-113. Wnt3a overexpression reduced
proliferation compared to vector only (LZRS) control, as measured by
BrdU incorporation (Figures 5A and 5B). WNT inhibition by treatment
with either GNF- 6231 (Figure 5A) or by C-113 (Figure 5B) for 24 hours
reduced the anti-proliferative effect of WNT3a overexpression. Similar
effects were observed in Sca1+ cells stably expressing Axin2, a WNT pathway
negative regulator (data not shown). We were unable to confirm the
effects WNT pathway on proliferation of Sca1+ cells in-situ because of
technical challenges in identifying Sca1 or c-kit expressing progenitors
in the heart by immunostaining. Interestingly, GATA4 transcription
factor, which is expressed by early differentiating cardiomyocytes
[34], was localized to the nucleus in more cardiomyocytes in the
infarct border zone of GNF-6231 treated hearts compared to control
(Figures 5C and 5D). Taken together, these observations suggest that
the proliferating cells in the GNF-6231 treated myocardium may include
myogenic progenitors.
WNT inhibition enhances survival of cardiomyocytes
We next asked whether the preservation of myocardial function and
smaller infarct size in GNF-6231 treated animals could be accounted for,
at least in part, by an effect on myocyte death or survival. Previous studies
have shown that WNT inhibition by sFRP2 improves cardiomyocyte
survival in the MI model [35] and in culture, specifically by binding
to WNT3A and blocking its pro-apoptotic signals [36]. Terminal
deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL)
was performed to detect cell death in situ. Co-immunostaining for
TUNEL and the cardiomyocyte marker cTnI showed that in the infarct
border zone, there were significantly lower percent TUNEL+ (apoptotic
or necrotic) cardiomyocytes in the GNF-6231 treated hearts compared to
vehicle-treated (2.03 fold lower in GNF-6231 treated ventricles; *p=0.022;
Figures 6A-6C). For further verification of the positive effect of WNT
inhibition on cardiomyocyte cell survival in vitro, cell survival and cell
death assays were performed on isolated cardiomyocytes. In the mouse
cardiomyocyte cell line, HL-1, cell viability was assessed by measuring
metabolic activity resulting in reduction of 3-(4,5-dimethylthiazol-2-
yl)-2,5- diphenyltetrazolium bromide—MTT— to insoluble formazan
(MTT assay). Treatment with 50 ng/mL mouse recombinant WNT3A
over 48 hours significantly reduced survival of these cells (by 16.4% over
control; *p ≤ 0.05; Figure 6D), which could be reversed by treatment
with WNT inhibitor, C-113 (18.78% increase over WNT3A treatment;
*p ≤ 0.05; Figure 6D). Inhibition of WNT pathway in these cells by
C-113 was determined by real-time RT-PCR for WNT target gene Axin2
expression (Supplementary Figure 4B). Since BrdU incorporation assay
had indicated no effect on proliferation of these cells by WNT pathway
modulation (Supplementary Figure 4C), we concluded that the effect on
cell viability was exclusively due to reduction in cell death. To further
confirm these findings, we used human iPSC-derived cardiomyocytes
(iCell®
Cardiomyocytes2
; Cellular Dynamics International, Madison, WI).
This highly pure population of cardiomyocytes expresses cardiomyocyte
markers, cTnI (Supplementary Figure 4A), and Sarcomeric Alpha
Actinin (Ref [37] and manufacturer’s datasheet), and beats in culture.
TUNEL assay with these cells showed that in the presence of oxidative
stress induced by 250 µM H2
O2
, treatment with recombinant WNT3A
significantly increased cell death (percent TUNEL+ cells 1.8 fold higher
over control; **p ≤ 0.01), whereas WNT inhibitor treatment reduced
cell death (by 3.6 fold over WNT3A treatment; ***p ≤ 0.001; Figures 6E
and 6F), further suggesting that enhanced myocyte survival and reduced
myocyte death contributed to the observed pro-reparative effect of postinjury
WNT inhibition.
WNT inhibition reduces type I collagen mRNA expression by
cardiac myofibroblasts
As discussed in the preceding sections, GNF-6231 treatment reduced
myofibroblast proliferation compared to vehicle-treatment (Figure 4I).
Since myofibroblasts are the major matrix synthesizing cells responsible
for scar formation, we tested whether WNT inhibition also affected
type I collagen synthesis by cardiac myofibroblasts. Primary αSMA+
myofibroblasts (Figure 7A) were generated from adult mouse hearts as
previously described [26]. Treatment of these cells with WNT inhibitor
(1 µM C-113) for 48 hours reduced Collagen1α1 gene expression (by
39.16% over control; *p=0.0251) as determined by qRT-PCR (Figure 7B).
These data suggest that WNT inhibition reduced pro-fibrotic effects in
the infarcted heart by modulating both the proliferation and the matrix
synthesis activity of cardiac myofibroblasts.
Discussion
Several studies have reported that canonical WNT signaling is temporally
increased after MI [4,5,38]. In this study we showed that temporary
systemic inhibition of the WNT/β-catenin signaling by blocking WNT
ligand secretion for several days post-MI prevented this post-MI tissue
WNT activation. Furthermore, therapeutic WNT inhibition following
infarct alleviated adverse cardiac remodeling, improved ventricular
function, and reduced infarct size. These findings corroborate published
reports regarding the positive effects of short term WNT inhibition with
small molecule pyrvinium [11,12] on post-injury repair. While our study
mirrored these reports regarding increased cell proliferation [11] and
reduced cardiac myofibroblast proliferation [12] in response to WNT
inhibitor treatment, we found, in contrast to these studies, that GNF-6231
did not affect endothelial cells (vWF+) proliferation, and that increased
survival of cardiomyocytes was a potential mediator of improved postinfarct
recovery. These differences may be due to the limited treatment
[11] and sequelae posed by toxicity of pyrvinium, blocking of both liganddependent
and ligand-independent WNT signaling, or confounding
effects of collateral inhibition of the Hedgehog signaling pathway by
casein kinase1α targeted by pyrvinium [39].
Our results also differed from the study in which WNT3A/5A
antagonist peptides were delivered via mini-osmotic pumps over 5 weeks
following MI [10]. Although we observed similar pro-reparative effects
on cardiac function, remodeling and infarct size, the effects on specific
cell populations were notably different. By contrast to the increase in
myofibroblast number and type I collagen synthesis, we observed a
reduction in αSMA+ myofibroblast proliferation and type I collagen
expression by myofibroblasts. Given the role of myofibroblast in scar
production in the heart and other organs, reduction rather than increase
in myofibroblast number and activity would be expected to contribute
to reduced scarring post-infarct. Effects on cardiomyocytes, progenitors
and other fibroblast populations were not studied [10]. These differences
in cellular effects of WNT inhibition may be attributed to incomplete
targeting of the WNT pathway through a subset of WNT ligands (e.g.
inhibition limited to WNT3A and WNT5A) [10], or extended treatment
over the entire MI repair process.
An important strength of our study was the utilization of a
therapeutically relevant small molecule, whose complete WNT inhibitory
activity on all ligand-dependent WNT signals, persisted at least up to
24 hours post-intravenous injection, allowing a daily injection regimen
(obviating the need to deliver biologic via mini-osmotic pump). this does
not pose toxic effects on other WNT dependent tissues. Additional finetuning
of the chemistry of the drug and dose or dosing regimen could
further improve the healing outcome by Porcupine inhibition. We build
on the finding that short term pharmacologic WNT inhibition improves
cardiac function and reduces adverse remodeling, by an expanded
investigation of the cellular mediators of this effect. Temporary WNT
inhibition post-infarct increased cell proliferation and cardiomyocyte
survival, and reduced myofibroblast proliferation and their matrix
synthesis activity in the heart. Examination of other fibroblasts marked
by expression of FSP1 or Vimentin, and vWF+ endothelial cells showed
no effect on proliferation of these cells by GNF-6231 treatment compared
to vehicle control. Whereas previous studies on pharmacologic WNT
inhibition have focused on specific cellular mediators of infarct pathology
(e.g.: on myofibroblast proliferation and activity, and neo-vascularization
[10]), our study includes a more comprehensive examination of the
cellular mediators of improved repair.
Early after infarct, WNT inhibition caused an increase in proliferation
of interstitial cells, particularly in the distal myocardium. The cardiac
cells that showed a proliferative response to GNF-6231 treatment
mostly excluded cardiomyocytes, endothelial cells and various stromal
populations, including αSMA+ myofibroblasts, and Vimentin+/Periostin+/
FSP1+fibroblasts. Interestingly, we discovered that WNT pathway activation
downregulated proliferation of isolated Sca1+CD31-
CD45-
CD117-
cardiac
progenitor cells, which are one of the tissue resident stem cells reported
to reside in the interstitial niche. WNT inhibition by treatment with two
mechanistically distinct WNT inhibitors, or via overexpression of Axin2
reversed the anti- proliferative effect of WNT activation in these cells.
This is in agreement with published reports of anti-proliferative effects of
recombinant WNT3A on side population progenitors, in vitro and in vivo
in the infarcted heart [30]. Our own work, and work by others in cardiac
injury and other injury models [11,32,40] report the WNT pathway as
a negative regulator of cell proliferation, particularly of stem/progenitor
cells. Although these data may appear incongruous with reports of WNT
being necessary for stem cell homeostasis and self-renewal in other adult
organs [41-43], and during development [44,45], our data support a
model in which WNT exerts multi-phasic, context dependent effects on
stem cells. For example, during heart development [46], just as in skeletal
muscle regeneration [29], a temporal regulation of WNT contributes to a
balance between stem cell proliferation and differentiation. Also, in the
now well-optimized and commercially used methods of cardiomyocyte
differentiation from iPSCs, a biphasic regulation of WNT activity is sought
in order to achieve optimal cardiomyocyte generation [47]. The observed
expansion of GATA4+ (i.e. newly differentiating) cardiomyocytes in the
infarct border zone provided in vivo support of the role of WNT inhibition
in enhancing neomyogenesis.
Our data also suggest an anti-fibrotic effect of WNT inhibition
following MI. We found that WNT inhibition reduced the number of
proliferating myofibroblasts in vivo, and also downregulated Collagen
I expression in cultured cardiac myofibroblasts. These results are not
surprising against the backdrop of numerous studies reporting that WNT
activation is a driver of fibrosis in heart [48] and many other forms of
tissue injury [49,50].
In addition to effects on cell proliferation and myofibroblast activity,
WNT inhibition also reduced cardiomyocyte cell death, which is the
major cause of the subsequent progression to heart failure [51]. This
observation aligns with published reports of pro-apoptotic effects
of WNT [30], and pro-survival effects of WNT inhibition [35,36] on
cardiomyocytes.
In this study, we focused exclusively on the effects of Porcupine inhibition
through the β-catenin-dependent arm of the WNT pathway based on the
significant body of literature suggesting a critical and complicated role—
both maladaptive [6,7,30], and in some cases pro-reparative [13,14,16]—
for this signaling cascade in infarct pathology. However, since GNF-6231
targets Porcupine, its effects independent of canonical WNT pathway
may also be important. Porcupine acyltransferase activity is specifically
targeted towards WNT ligands [20,22], and hence other pathways are
unlikely to be affected. However, Porcupine inhibition can also affect the
non- canonical arms of the WNT pathway.
There are reports implicating both the Ca2+/CAM Kinase and the JNK
arms of the non-canonical WNT signaling pathways [34,52] in infarct
pathology and repair. Hence, future studies investigating the effect of
inhibition of WNT ligand secretion on the non-canonical arms of the
WNT signaling pathways may provide a more complete picture of the
mechanisms mediating cardiac recovery by Porcupine inhibitor treatment.
Moreover, WNT/β-catenin pathway can be activated downstream of
ligand binding through cross-talks with other pathways such as TGFβ
[53,54]. The contribution of ligand-independent WNT pathway activation
in infarct pathology remains unclear. Development of a biocompatible
agonist of the WNT/β-catenin degradation complex (similar to pyrvinium,
but without the toxicity) would be useful in answering these questions.
That said, our data demonstrating the potential of short term WNT
inhibition in counteracting the key drivers of post-infarct LV dysfunction
and eventual failure—cardiomyocyte death and fibrosis—are clinically
significant since the current standard-of-care for myocardial infarct focus
mainly on thrombolytic and palliative interventions, and do not address
the ongoing disease progression driven by the initial infarct. Moreover,
in the context of complicated [5,9,13,16,32] and multifaceted roles of the
WNT pathway in infarct repair in the existing literature, our data may
speak to the potential of temporally regulated scalable pharmacologic
WNT inhibition in reconciling the discordant observations based on
genetic models [6,7,14] of WNT modulation. With additional details
on the mechanism-of-action and safety data emerging with continuing
studies, and ongoing clinical trials [23], GNF-6231 and the new class of
Porcupine inhibitors hold significant potential as effective therapeutics for
cardiac regeneration.
Funding
This work was supported by the Veterans Affairs Merit Award, NIH
grants R21EB019509-01A1, and 1R01GM118300 to PPY, and Vanderbilt
University Clinical and Translational Science Award [Grant number UL1
RR024975-01] from National Center for Research Resources (NCRR)/
National Institute of Health (NIH), and philanthropic funds to PPY; and
the American Heart Association Predoctoral Fellowship [3PRE16080004] to DB.
Acknowledgments
We would like to acknowledge Dr. Antonis K. Hatzopoulos for providing
the TOPGAL mice, Dr. Ethan Lee for C-113, the Genomics Institute of
Novartis Research Foundation for GNF-6231, the Translational Pathology
Shared Resource (TPSR) at Vanderbilt University Medical Center for aid
in specimen preparation for histology, and Dr. Bin Li and Dr. Caressa
Lietman for their constructive criticism.
Author Contributions
DB and PPY designed the study. DB, SS, PJ, JA, JL and JLH performed
experiments and collected data from experiments. DB, SS, JL and PPY
analyzed the data, IF, JL and JLH provided reagents and helped in data analysis
and provided conceptual advice. DB, JL, JLH and PPY wrote the manuscript.
Conflicts of Interests
JL and JLH are employees of the Novartis Research Foundation. PPY
is listed as inventor for a WNT inhibitory topical therapeutic.
