Stem Cell Development
The development of adipocytes in mice and humans follows a
well-defined pathway that commences with a common pluripotent
mesenchymal stem cell (MSC), ie., adipogenesis [1]. The early steps of
the pathway leading to the generation and the commitment of MSCs to
an adipocyte lineage are unknown. Hypothetically, the determination of
the fate of MSCs occurs early in cell differentiation (“commitment”) and
involves the interplay of intrinsic (genetic) and environmental (local and
systemic) conditions that ultimately define the fate of the cell. Factors
that determine MSC proliferation and differentiation also govern early
adipocyte development and function. Currently, little is known about
this process; from MSC-to-preadipocyte differentiation. However,
the steps governing the transition from preadipocyte to adipocyte
differentiation are not well defined (Figure 1). During adipogenesis MSCs
or preadipocytes differentiate into lipid-laden adipocytes [2]. Ox-HDL
increases adipogenic properties with a marked effect on the last step of
adipocyte-terminal differentiation and release of adipokines including
20- HETE and Ang II.
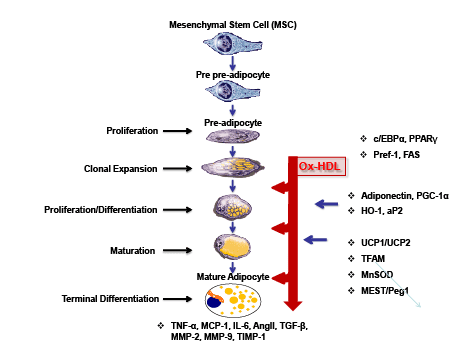
Figure 1: Schematic presentation of MSCs giving rise to adipocyte differentiation.
MSCs can differentiate into adipocytes when placed in the adipogenesis medium in vitro. Various adipokines including Ang II, Leptin, TGFβ, VEGF, FGF,
HGF, TNF, Adiponectin, MMP-2, MMP-9 and IGF-1 are secreted from adipocytes. Particular molecular events accompanying each stage of differentiation
are indicated to the right, with the imprecise interval in each stage reflected as indicated.
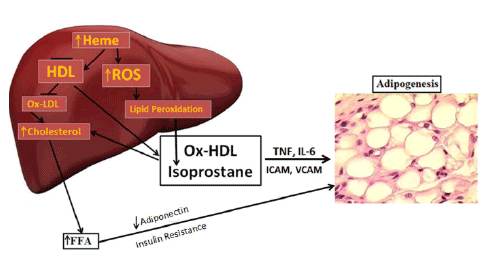
Figure 2A: HO-1 decrease ratios of LDL/HDL in obese mice treated with CoPP, obese mice display high levels of LDL, while treatment with CoPP,
for 4 weeks decrease LDL, that is reversed by inhibition of antioxidant HO-1
MSCs were initially identified in postnatal human bone marrow
and have been used to model differentiating mesoderm. It is believed
that the MSCs give rise to a common early precursor (pre-adipocyte,
Adipoblast), which, in turn, develops into the committed white and
brown preadipocyte that under appropriate stimulatory conditions,
differentiate into mature adipocytes of different types [3]. The transition
from preadipocyte to adipocyte involves four stages: growth arrest,
clonal expansion, early differentiation and terminal differentiation [4].
Adipocytes regulate glucose homeostasis [5] and adipocyte dysfunction
results in the secretion of decreased levels of adiponectin and decreased
glucose uptake, leading to insulin resistance [6].
Obesity and Ox-HDL
Obesity is also linked to the metabolic syndrome, which is associated
with a dyslipidemic profile that includes hypertriglyceridemia and low
plasma high-density lipoprotein cholesterol (HDL-C). Accumulated
evidence suggests that HDL enhancement plays a beneficial role in
maintaining glucose homeostasis via insulin dependent and independent
pathways. Low Density Lipoprotein Cholesterol (LDL-C) and HDL-C
levels have become the accepted biomarkers in the evaluation of the risk
of CVD, CAD, and even CKD [7,8]. Recent studies have suggested
that HDL function is more important than total levels of HDL and that
remodeling and dysfunction likely contribute to increased risk of CVD,
CKD, and CRS.
High fat diets increase LDL and glucose levels [9] which are both
reversed by an increased expression of the antioxidant gene, heme
oxygenase (HO-1). In another model of high fat (HF) diets in hypertensive
rats, LDL is increased and this is prevented by induction of HO-1 by a
number of cobalt compounds including cobalt protoporphyrin [10].
Similar observations are described for male and female mice [11]. These
observations are attributed to increases in ROS in adipose tissue and
liver that may involve increases in Ang II and 20- HETE, which are
major sources of ROS [12]. Deletion of angiotensinogen in hepatocytes
markedly decreased blood pressure [13]. Angiotensinogen has been
synthesized by 3T3- F442A cells and hydrolyzed to ANG l and ANG
II in adipocytes [14], and its deletion from adipose tissue resulted
in a decrease in blood pressure elevation in obese mice [15]. In
another study, increases in antioxidants decrease the Ang II-mediated
increase in ROS [16- 18]. These reports suggest that targeting the Ang II
system may have therapeutic value. The increase in ROS is considered a
contributing factor in Ox-LDL [19] in contrast to an increase of HO-
1, which inhibits atherogenesis [20] and atherosclerotic lesion in LDL
receptor (-/-) mice [21], reviewed in [22].
Dysfunctional HDL can result from both free radical attack and
oxidation of ‘good’ HDL, leading to Ox-HDL (‘bad’ HDL) [23-25].
Lipids and lipoproteins are the primary targets of free radical damage
[26], which results in lity and CVD and cardiac events.
Process of MSCs differentiation to Adipocytes
HO-1 effect Plasma LDL and HDL
We believe that levels of antioxidants will change the ratio of LDL and
HDL in mice. As shown in figure 2A and 2B, the ratios of plasma LDL
and HDL is significantly higher in obese mice than in lean mice (0.41
+ 0.15 vs 0.05 + 0.02, *p<0.05). An increase of HO-1 and antioxidant
properties [12,39] by CoPP decreased the ratio (0.15 + 0.01 vs 0.41 +
0.15, *p<0.05). Inhibition of HO-1 and increase of antioxidant by SnMP
blocked the effect of CoPP on obese mice.
The Effect of Ox-HDL and Isoprostanes on Adipogenesis
We examined the levels of LDL to HDL in mice treated with CoPP, which
increases HO-1-derived bilirubin levels. Since obesity is associated with a
decrease of antioxidants, we propose that this will result in an increase in
levels of Ox-HDL as Ox-HDL is increased in cardiac events. We examined
the effect of Ox-HDL and isoprostanes on adipogenesis in the human
adipocyte by measuring Oil Red O stained lipid droplet area after 10
days of treatment (Figure 3). The level of Oil Red O stained lipid droplets
increased after treatment with Ox- HDL, isoprostanes, and a combination
of the two. Quantification of Oil Red O stained cells showed an increase in
lipid droplets in the presence of both Ox-HDL and isoprostanes compared
with control p<0.05 and Ox-HDL. This effect proved to be synergistic,
p<0.05 (Figure 3). These results were confirmed in mice (results not shown).
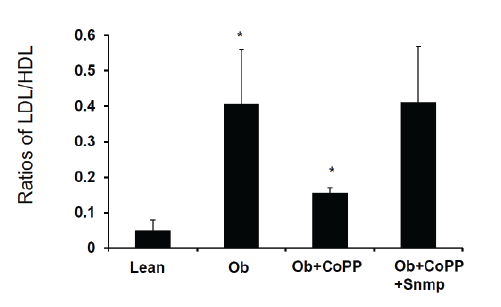
Figure 2B: HO-1 decreases ratios of LDL/HDL in obese mice treated with CoPP, obese mice display high levels of LDL, while treatment with CoPP,
for 4 weeks decreases LDL, that is reversed by inhibition of antioxidant HO-1.
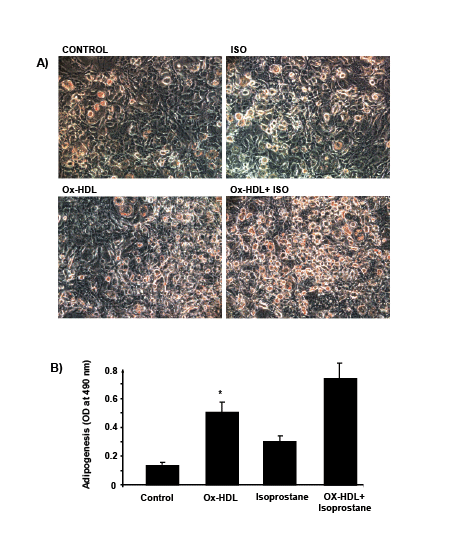
Figures 3: Adipogenic effect of oxidized HDL and isoprostane on MSC-derived adipocytes. Adipogenesis from human MSC was detected by Oil Red
O staining and absorbance was measured as described [37,39]. *p<0.05 versus control.
Figure 4 is a schematic that shows the release of the inflammatory
cytokines IL-6, and TNF and ROS. ROS increases lipid peroxidation
with increased levels of Ox-HDL, LDL and isoprostane. Excess heme,
needed for adipocyte differentiation and terminal differentiation, also
increases ROS. Hyperglycemia in the obese will also increase the levels of
ROS (Reviewed in [12]). With the down regulation of HO-1 in obesity,
heme catabolism is decreased. ROS targeting adipocyte stem cells and
hypertrophy occurs in several animal models of obesity which leads to an
increase of inflammatory adipokines, a decrease in adiponectin, liver and
muscle fat deposit and insulin resistance.
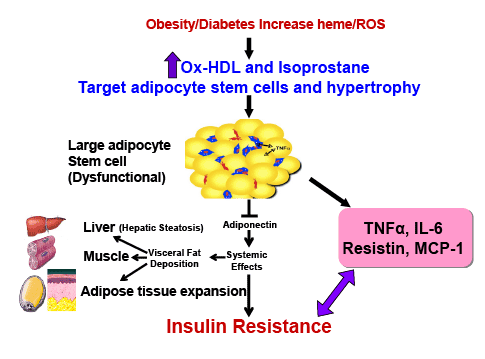
Figure 4: Schematic representing the increase in ROS by high fat, glucose or excessive heme levels that in turn increase the generation of oxidized
HDL and isoprostane. Enlargement of adipocytes causes alterations in the secretion of adipokines. Increased adipocyte size can lead to deleterious
alterations in insulin sensitivity caused by a decrease in adiponectin secretion and the induction of inflammatory mediators.
This review demonstrates that Ox-HDL and isoprostane exert marked
increases in adipogenesis in human adipocyte stem cells. Ox-HDL is
associated with an increase in adipocyte expansion and adiposity and, as
such, is a determinant of obesity and its related disorders. There are several
ways in which Ox-HDL can be formed. One way is during the process of
differentiating adipocytes. This process begins with a high food intake,
early hyperglycemia occurs resulting in an increase in cellular heme due to
a decrease in the levels of HO-1 (reviewed in [12]). Heme is a pro-oxidant
and a source of ROS which contribute to an increase in NO uncoupling
by iNOS induction. The induction of iNOS causes the formation of
peroxynitrite which is responsible for lipid peroxidation and inhibition
of protein and enzyme function and increased Ox-HDL levels. A prime
example is a decrease in the levels of HO-1 which, in turn, decreases
bilirubin levels. Bilirubin is a potent antioxidant and patients with elevated
bilirubin levels display a lower risk of cardiovascular disease and have
higher levels of HDL (reviewed in [31]).
There are a number of mechanisms by which obesity increases the levels
of Ox- HDL. These occur during the process of differentiating adipocytes
that requires glucose, which is a major source of ROS. Furthermore,
myeloperoxidase is responsible for generating excessive levels of ROS [32]
with a resultant increase in lipid peroxidation which converts LDL and
HDL to oxidized products with an expansion of adipogenesis.
We and others have shown that an excess of heme in adipocyte stem
cells and in the fat of obese mice is necessary in order for adiposity [11,33-37].
Therefore, increased heme levels in obese subjects, is a major source of
ROS, contributing to lipid peroxidation and production of Ox-HDL and
Ox-LDL. Additionally, hemoglobin influences LDL and HDL in obesity
and diabetes. Hemoglobin increases the levels of proinflammatory HDL,
in other words, increases the oxidation of HDL. We believe that HDL
dysfunction is not the cause of adipogenesis, but it is the oxidation of the
HDL itself [38].
Obesity is a growing epidemic in the United States as well as worldwide.
Many of the cardiovascular complications associated with obesity are, in
part, due to dysfunctional adipocytes and endothelial damage. Several
clinical conditions such as diabetes mellitus and obesity, are characterized
by both increased inflammation and oxidative stress, and are associated
with increased risk of cardiovascular complications. An increase in OxHDL
negatively correlated with adiponectin levels in morbidly obese
subjects (unpublished data). Thus, HDL and Ox-HDL may prove of
particular relevance, in the maintenance and regulation of cardiovascular
health and as targets for the prevention of cardiovascular events.
In conclusion, this communication suggests that the novel finding
that Ox-HDL and isoprostane act at the three points presented in figure
1, and that it appears that Ox-HDL enhances adipogenesis and/or the
recruitment of stem cells in adipose tissue, and increases the adipogenic
lineage and exacerbates obesity and the metabolic syndrome. In support
of this conclusion, isoprostane , another oxidant found in the plasma of
obese subjects increases adipogenesis and, with Ox-HDL, synergistically
increases adipocyte stem cell proliferation, differentiation and hypertrophy.
Thus Ox-HDL function, due to its adipogenic effect on adipocyte stem
cells, should be re- evaluated to address the metabolic derangements
associated with the metabolic syndrome.
Acknowledgements
This work was supported by National Institutes of Health grants
HL55561, HL34300, HL 109015, The Brickstreet Foundation and The
Huntington Foundation (NGA, JIS). We thank Jennifer Brown for her
outstanding editorial assistance in the preparation of the manuscript.
Article Information
Aritcle Type: Short Communication
Citation: Peterson SJ, Vanella L, Bialczak A,
Schragenheim J, Li M, et al. (2016) Oxidized HDL
and Isoprostane Exert a Potent Adipogenic Effect on
Stem Cells: Where in the Lineage? Cell Stem Cells
Regen Med 2(1): doi http://dx.doi.org/10.16966/
2472-6990.109
Copyright: © 2016 Peterson SJ, et al. This is an
open-access article distributed under the terms
of the Creative Commons Attribution License,
which permits unrestricted use, distribution, and
reproduction in any medium, provided the original
author and source are credited.
Publication history:
Received date: 23 Dec 2015
Accepted date: 21
Apr 2016
Published date: 27 Apr 2016
