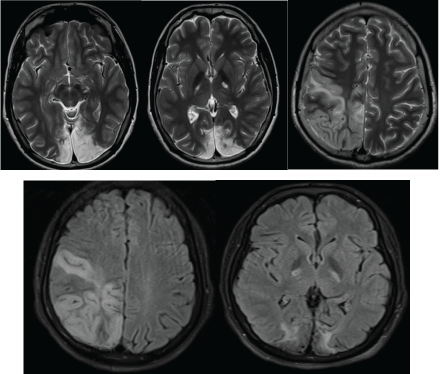
Figure 1: T2 and FLAIR hyper-intensity involving both white and grey matters of the bi-occipital regions, the right temporo-parietal region and the posterior limbs of the internal capsules.

Mahmoud M1* Ben Ammar M1 Kraoua I2 Drissi C1 Klaa H2 Ben Youssef Turki I2 Ben Hamouda M1
1Department of Radiology, National Institute Mongi Ben Hamida of Neurology, Tunis El Manar University, Tunis, Tunisia*Corresponding author: Mahmoud M, Department of Radiology, National Institute Mongi Ben Hamida of Neurology, Tunis El Manar University, Tunis, Tunisia, E-mail: mahmoud.maha@gmail.com
A 15-year-old boy, with a history of psychomotor retardation which began at the age of two years (with a retardation of walk which had started at the age of 20 months) and continued steady until the age of 13 years, presented in 2012 with focal epilepsy, further walk troubles and headaches. The child’s symptoms had previously been followed by a neurologist, who prescribed a medical treatment, unreported by the family
The magnetic resonance imaging (MRI) realized in May 2012 showed cortical and subcortical hyperintensity in both occipital lobes with a gyral enhancement and restricted diffusion in the right lesions. He received anti-epileptics and vitamin therapy with no recovery
In June 2015, he developed an ataxia and recurrence of focal epilepsy. The lumbar puncture revealed an elevated lactate ratio. MRI and MRspectroscopy imaging were undertaken, seeking a metabolic neuropathology.
The MRI revealed a wide signal abnormality, type of T2, FLAIR and diffusion hyperintensity involving the cortical and white matters of the right fronto-parietal regions and the occipital lobes (Figures 1 and 2). ADC cartography, showed a heterogeneous map, with regions of elevated ADC and some others with lowered ADC; these latter areas correspond with cytotoxic edema (Figure 2).
Figure 1: T2 and FLAIR hyper-intensity involving both white and grey matters of the bi-occipital regions, the right temporo-parietal region and the posterior limbs of the internal capsules.
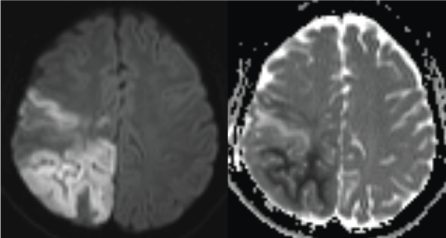
Figure 2: Diffusion B1000 and ADC cartography shows heterogeneous map with regions of increased ADC (green arrow) and regions with restricted diffusion (red arrow). These latter correspond with ischemic regions.
Proton-MR-Spectroscopy showed an elevated lactate ratio in both normal and pathological regions and in cerebrospinal fluid (CSF) with decreased N-acetylaspartate (NAA) ratio in pathological regions (Figure 3).
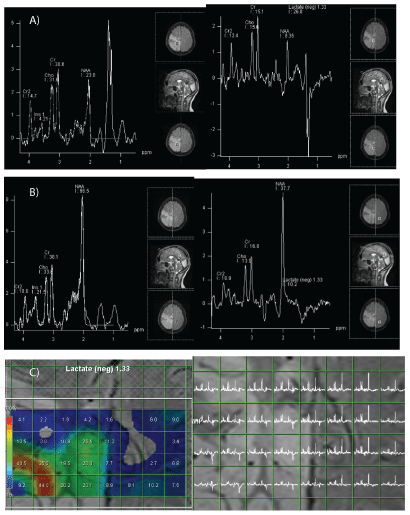
Figure 3: Spectroscopic study.
A: AMbnormal areas: elevated doublet of lactate at 1.33 ppm (red arrow) which is inverted on the spectrum with long echo-time (green arrow). NAA/Creatine ratio is reduced.
B: Morphologically normal areas: doublet of lactates at 1.33 ppm, which is positive on short echo-time and negative on long echo-time spectroscopy.
C: Spectrum cartography (at long echo-time) showing lactates peaks in both pathologic and morphologically normal areas.
In retrospective review with the results of a previous lumbar puncture (elevated lactate ratio), a metabolic pathology was suspected, and more precisely mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke like syndrome (MELAS), which can be associated with these typical imaging features.
MELAS syndrome is one of the most frequent maternally inherited mitochondrial disorders. It affects selectively the nervous system and muscles [1]. Diagnostic criteria of MELAS syndrome were published in 1992, and include the following criteria:
Stroke-like episodes are one of the most frequent features that occur in the MELAS syndrome rising as high as 84%-99% in affected individuals. Symptoms are partially reversible aphasia, cortical vision loss and motor weakness [2].
Dementia is also a frequent feature of MELAS syndrome, occurring in 40%-90% of affected individuals. Neurological dysfunction and cortical injuries due to stroke-like episodes are both incriminated in the occurrence of dementia [2].
Seizure occurs in 71%-96% of individuals affected by MELAS syndrome. It can occur independently, or as a manifestation of stroke-like episodes [2].
Furthermore, recurrent headache, hearing impairment and peripheral neuropathy are another common manifestation of MELAS syndrome.
Brain MRI is the most useful imaging technique to explore and reveal the most frequent imaging features of MELAS syndrome which includes [1-3]:
Stroke-like lesions, the basal ganglia calcifications and brain atrophy.
Stroke like lesions (SLL) are multifocal signal abnormalities in the cortical grey matter that do not match with vascular territory, but whose signal evolution looks like stroke.
In the acute phase, MRI shows high signal intensity in T2 and FLAIR weighted imaging of both grey and white matters, and hypo signal intensity on T1WI.
DWI and ADC sequences may show restricted diffusion in the SLL areas with a variable ADC. In fact, early reports showed an elevated ADC in stroke-like lesions due to a vasogenic edema, however recent reports showed that a cytotoxic edema can occur during acute phase of stroke-like episode, and may show a decreased ADC or mixed areas of high and low ADC. This is mainly due to different levels of mitochondrial impairment [24].
Spectroscopy findings are not specific to MELAS. It can show a decrease in N-acetylaspartate (NAA) that reflects a loss or impairment of neurons and an increase in lactate that reflects anaerobic metabolism. In addition, the lactate peak may be found in normal-appearing areas on MRI, suggesting and supporting the mitochondrial cytopathy theory [1,4].
In the sub acute phase, MRI findings are compatible with cortical laminar necrosis.
Cortical atrophy and symmetric basal ganglia and thalamic calcifications are also common imaging findings during the evolution of the disease.
The differential diagnosis should include [1]:
The rarity of MELAS syndrome and the complexity of its clinical features render it difficult to diagnose. A radiologist has to think MELAS in a patient with acute “stroke-like” cortical lesions that cross usual vascular territories and fluctuate through time. DWI and spectroscopy (in both pathologic and uninvolved brain and in CSF) are helpful tools providing information to obtain an accurate diagnosis.
Download Provisional PDF Here
Article Type: Case Report
Citation: Mahmoud M, Ben Ammar M, Kraoua I, Drissi C, Klaa H, et al. (2016) MELAS MR and Protonic-MR-Spectroscopy Findings: A Case Report. Pediatr Neonatal Nurs Open Access 2(3): doi http://dx.doi.org/10.16966/2470-0983.113
Copyright: © 2016 Mahmoud M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access