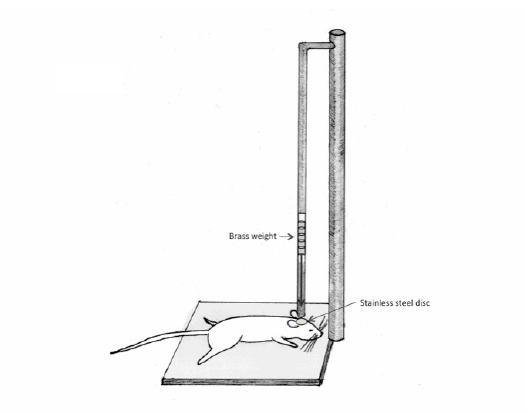
Figure 1: Schematic illumination of a weight drop model of TBI. The device to make this model consists of a column of brass weights, which ultimately falls freely via gravity.
Michael Galgano Thomas Russell Sandra Mc Gillis Gentian Toshkezi Lawrence Chin Li-Ru Zhao*
Department of Neurosurgery, State University of New York, Upstate Medical University, Syracuse, New York, 13210, USA*Corresponding author: Li-Ru Zhao, Department of Neurosurgery, State University of New York, Upstate Medical University 750 E, Adams Street, Syracuse, NY 13210, USA, Tel: 315- 464-8470; Fax: 315-464-5504; E-mail: ZHAOL@upstate.edu
Traumatic brain injury (TBI) continues to be a significant problem affecting many individuals on a daily basis. There are various subtypes of traumatic brain injuries ranging from life-threatening cerebral edema to the injuries not accounted for by current radiographic imaging. To understand the pathological features of TBI and evaluate potential therapeutic strategies for TBI, various animal models have been created and characterized. Each of the animal models is aimed to mimic a certain type of clinical TBI. In this review article, we critically evaluate the various types of animal models for TBI, and describe which patient populations these models are intended to represent.
Mild traumatic brain injury bears the highest representation of TBI in the United States. Chronic traumatic encephalopathy (CTE) is the longterm sequelae of such recurrent mild brain injuries. We particularly emphasize the animal model for CTE in this article because research in this arena tends to significantly lag behind societal awareness of this ever-growing problem amongst our nation’s athletes. Repetitive traumatic brain injuries in humans have the potential to cause significant neurobehavioral and psychiatric disturbances years after the inciting events, as the injuries accumulate over time. Postmortem examination of brains from the patients who experienced repetitive traumatic brain injuries displays pathological profiles of CTE. Creating animal models for CTE have only just begun. The models that culminate in the animal-equivalent of CTE in humans remain to be developed in future studies.
Chronic traumatic encephalopathy; Traumatic brain injury; Animal model
Traumatic brain injury (TBI) represents a wide spectrum of various acquired insults to the central nervous system. TBI is seen in a variety of forms, from local and mild injury to diffuse and severe. Mild brain injuries may cause subtle alterations of consciousness almost unnoticeable to the patient, while severe injuries have the capacity to be incompatible with life. Clinical presentations vary from an acute nature immediately after the impact to a more insidious onset due to cumulative effects over time.
TBI is a growing problem in the United States. It is estimated that approximately 1.7 million people sustain a traumatic brain injury annually, and of those individuals, over 50,000 die. TBIs may occur across all ages and populations. In fact, almost half a million emergency room visits per year for TBI are represented by children aged 0-14 years.
Elderly individuals aged 75 years and older remain the most susceptible population to TBI-related hospitalizations and deaths. Falls and motor vehicle accidents continue to be the main etiological factors for TBI [1]. Sporting activities involving rapid acceleration/deceleration as well as collisions also are a great risk factor for different types of TBI.
Mild traumatic brain injury (mTBI) actually represents the majority of TBI in the United States. Well over 1 million people suffer a TBI annually without any loss of consciousness, or subsequent hospitalization [2] .Many of these mild traumatic brain injuries are loosely defined as concussions. It is evident that although concussions do not typically represent a significant acute event in and of themselves, the culmination of repetitive mild brain injuries is now being brought to the attention of the public eye, most notably through American sports. A study of high school athletes found that individuals involved in all sports were at risk for concussions with boys’ football the highest at 63% of the total, followed by wrestling (10%), soccer (6%), and the remainder in basketball, softball, baseball, field hockey, and volleyball [3,4]. In a prior prevalence study, it was found that nearly 17% of retired professional boxers exhibit characteristics of chronic TBI [5]. Public interest has peaked in the past few years given the number of high profile athletes that have committed suicide, and/or presented early onset of neurodegenerative disease and were subsequently found to be suffering from chronic traumatic encephalopathy (CTE). Over the last few years, there has been a rapid increase in awareness of CTE within the society as well through media outlets.
Unfortunately, translational research in this arena continues to lag behind the societal awareness of CTE [6].
Treatment for TBI alters greatly based on the location and severity of the insult. Severe diffuse brain injury may require an extensive surgical intervention such as bilateral decompressive craniectomies; in order to release the elevated intracranial pressure, the majority of both frontal, temporal, and parietal bones of the calvarium is removed. Other less severe forms of traumatic brain injuries are approached with more conservative treatment modalities, such as therapies addressing the subtle cognitive, motor or other neurologic deficits patients may be experiencing. The subject of CTE, however, is proving to be a difficult subtype of traumatic brain injury to provide an adequate treatment regimen for, as many of the victims are emerging years after their primary insults. In order to gain a better understanding of CTE, and how it differs from other types of brain injuries, we must start with small-scale animal models that are both reproducible and valid. As we will further discuss, a multitude of TBI animal models have been created for diffuse injuries, blast injuries, and controlled cortical impact. Unfortunately, models paralleling repetitive mild brain injury culminating in the equivalent of an entity such as CTE are lacking. The aim of this review is to give an introduction into the various TBI subtype models, and propose a model that would ultimately enable us to better understand the pathophysiology and treatment of CTE.
Various types of TBI animal models exist, each created to address representations of patient subpopulations. The most common TBI animal models include non-impact head acceleration, weight drop, blast wave, fluid-percussion, controlled cortical impact (CCI), and repetitive mild traumatic brain injury / chronic traumatic encephalopathy (rmTBI/CTE). Direct dynamic brain injuries can be sub classified as impact or nonimpact / acceleration head injury models [7]. These can be further divided into models where the head is constrained in a single plane, or is allowed to move freely [6].
Many of the most serious head injuries we see in clinical practice involve rotational acceleration of the brain within the rigid skull. This rapid acceleration of the brain is one of the main etiological factors associated with a diffuse brain injury pattern. The rotational mechanism involves shearing forces, which ultimately has the potential to cause a devastating clinical picture of diffuse axonal injury. Non-impact head acceleration animal models have been used in an attempt to replicate such injuries. A primate model has been generated whereby a pneumatic shock tester creates a controlled single rotation of the head, without any impact. The rotational acceleration has a biphasic pattern, with a long acceleration and short deceleration phase [8]. Later work by Margulies et al. showed diffuse axonal injury patterns in primates [9]. Smith and coworkers developed a head acceleration miniature swine model. The head is secured to a pneumatic actuator via a snout clamp. Once the pneumatic actuator is triggered, linear motion is produced which is subsequently converted to angular motion. Device activation rotates the head rapidly at a predetermined angular excursion, causing a diffuse axonal injury [6,8,9].
The advantages of non-impact head acceleration models are their applicability to clinical cases involving rotational force, but due to differences in brain mass between animals and humans (specifically mice) [10,11] the replicability and feasibility of the models is reduced.
One of the most widely used models to replicate diffuse TBI is Marmarou’s weight drop acceleration-impact model [12]. The preferred animal has been the adult rats, weighing approximately 350-400g. This model involves head impact followed by prolonged loading into a foam pad under the animal [12]. The device consists of a column of brass weights, which ultimately falls freely via gravity (Figure 1). The height from which they fall can be manipulated through a plexi glass tube. Once the animal is anesthetized, a midline incision is made exposing the underlying skull. A stainless steel disc is then rigidly attached with cement to the skull in a central location between the bregma and lambda. The animals are then placed onto a foam bed. The impact is generated by the free falling weights falling directly onto the cemented stainless steel disc [7].
This particular model is used to represent diffuse brain injuries in humans caused by falls and motor vehicle accidents. It has been shown to reproduce neuronal and axonal pathology that is scattered throughout the cerebrum and brainstem. The impact triggers a significant neuroinflammatory response within the intrathecal compartment. This ultimately leads to neurological impairment and breakdown of the blood brain barrier [13]. This model can be utilized in such a way that the TBI can be graded as mild, moderate, or severe. The grading can be manipulated based on changing pre-impact parameters such as varying the weights, the weight height, as well as post-impact implementation of secondary insults such as hypoxia and hypotension [14,15]. The injuries sustained with Marmarou’s weight drop model tend to be more severe than that compared to a similar impact mass and velocity to the freemoving head [12].
Figure 1: Schematic illumination of a weight drop model of TBI. The device to make this model consists of a column of brass weights, which ultimately falls freely via gravity.
The advantages of Marmarou’s weight drop model include: the ability to represent graded forms of diffuse TBI, ease of use and manipulation, replicability and well-studied neuropathology [12]. The disadvantages include: re-hits [16], skull fixation [12], and the use of anesthesia [17–23]. A more comparable model would entail similar weight and impact to a free-moving head, as is often encountered in humans involved in falls and motor vehicle accidents.
Gurdijan et al. demonstrated in the 1950’s that traumatic brain injuries could in fact take place via direct pressure loading without global head acceleration / deceleration forces being present [10,11]. Traumatic brain injuries from improvised blast explosives become a signature injury involving military personnel. The blast wave- induced brain injuries are typically caused either from flying fragments /shrapnel, or from shock waves. Animal models can be employed by exposing the subjects to explosives, weapons, or shock tubes [24]. However, field experiments can be strongly influenced by the environment. Shock tubes do appear to be easier to control, and therefore have a higher rate of reproducibility. Blast experiments do tend to come at a high cost, and as a result, experimenters are now employing numerical simulation techniques such as the finite element method. This is now being used as an important adjunct to experimental studies, as it allows the researcher to study the effect of complex brain/blast wave interaction with the entire brain [24].
The typical blast exposure model consists of an anesthetized animal first being fixed in a holder, which essentially prevents movement of their body in response to the blast. Pressure waves are then generated by either detonation of a plastic explosive or compressed air within the closed end a cylindrical metal tube, directed towards the head (Figure 2) [7]. Blast wave models have been known to cause behavioral deficits, diffuse axonal injury [25], white matter injury [26], neuro degeneration and glial activation [27].
The advantages of the blast wave model include its highly clinical relevance to returning veterans and its reproducibility. The disadvantages of the blast wave model include high price and the complications of the model [28,29]. The blast injury model created by high velocity ballistic penetration or stub injury may present with intraparenchymal hemorrhage along the trajectory of the first impact. This model also may reproduce other components with cerebral inflammation, breakdown of blood brain barrier, or other components related to CTE.
The fluid percussion model is able to produce brain injuries by the rapid injection of fluid pulse volumes into the cranial cavity through a craniotomy defect, directly onto intact dura [30]. The clinical correlate of this model is cerebral concussion [31]. To make this model, the anesthetized animal is placed in a stereotaxic frame, and their scalp and temporal muscles are then reflected out of the direct surgical field. The craniotomy may be made with or without a contralateral skull opening [32]. A plastic cap is then cemented into place. The fluid percussion device consists of a plexiglass cylindrical reservoir, which is filled with sterile saline. One end of the reservoir has a transducer.
This is mounted and attached to a tube, which is connected through a plastic fitting to the plastic cap cemented on the animals’ skull. A pendulum strikes at the opposite end of the reservoir. This ultimately generates a pressure pulse delivered to the intact dura. This causes deformation of the underlying brain (Figure 3) [7,33]. Varying magnitudes of fluid percussion can be undertaken in an attempt to produce graded neurological injury [14]. Subtleties in the technique and location of fluid percussion can aid in representing different forms of TBI. A midline injury tends to cause a “diffuse” injury, whereas lateralizing the impact causes a more focal injury pattern [14]. Most studies to date demonstrate that fluid percussion models induce a moderate level of brain injury. However, it is rather difficult to quantify the impact. In fluid percussion models, there tends to be brain “disruption,” as opposed to brain “destruction.” Therefore, the injury to the brain occurs in a broad range, and a large sample size may be necessary to detect significant differences among the groups [14].
The fluid percussion model manifests pathologic features of TBI such as axonal injury [34], neuroinflammation [35], neuronal loss and glial activation [36]. This model also leads to impairments in spatial learning and memory [34], cognitive deficits, and deficits in motor learning [37]. One advantage of the fluid percussion model is that it is a commonly used model with well-defined pathology and neurobehavioral deficits [38].
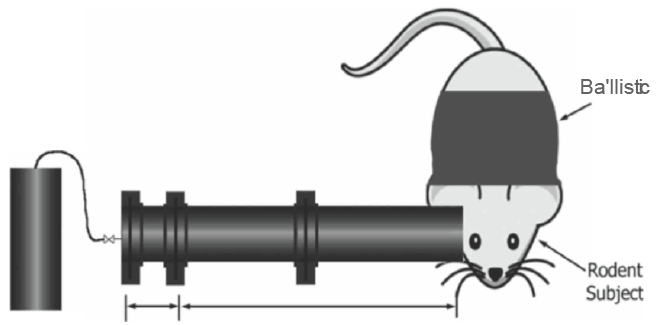
Figure 2: Schematic diagram of a blast wave model of TBI. The compressed gas in the driver section is released as a blast wave to the head.
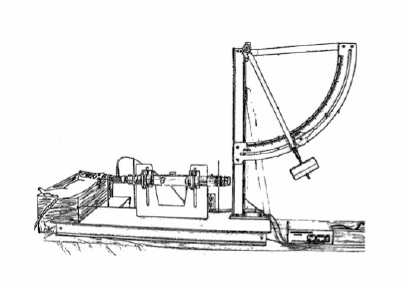
Figure 3: Schematic representation of a fluid percussion model of TBI. Brain injury is produced by a sudden force induced rapidly injecting fluid volumes onto the intact dual surface through a craniotomy.
The disadvantages of fluid percussion models include the broad range of injury [14], the reliance on operator skill for the pendulum, and concerns in reproducibility due to variability of friction within the apparatus [39], craniectomy [40] and anesthesia [17– 23,41].
The most commonly employed technique for a controlled cortical impact (CCI) TBI model entails the use of a mechanically actuated piston. The velocity and depth of the impact can be manipulated. Unlike the fluid percussion model, CCI is able to take advantage of the biomechanics of the brain injury, and better quantify the parameters and outcome. Force, velocity, tissue deformation, and the magnitude of tissue damage and subsequent functional impairment can be measured [14]. The typical CCI impactor is made of a rigid cylinder, mounted onto a crossbar. CCI models originally used a round impactor tip, whereas newer studies are using a flat or beveled tip [42]. The particular device we have used in our laboratory has the cylindrical flat-tipped impactor situated in a perpendicular fashion to the animal’s head, although other models demonstrate that it can be used in a horizontal fashion. After a craniotomy is performed on the animal, the pneumatically-driven impactor is set at predetermined parameters prior to impact (Figure 4). The CCI model produces a pronounced cortical contusion and subarachnoid hemorrhage, unlike many of the other models [43]. The deficits caused by CCI models have been found to mimic neurobehavioral and cognitive deficits, as they are typically seen after human TBI [14,44], such as depression, anxiety, impairment of spatial learning and memory, and multiple motor deficits [45].
The advantages of CCI models include the ability to directly control physical damage, reliable deficits [46], and no rebound concussive events (as seen in weight drop) [16].
The disadvantages of CCI models include craniotomies and clinical application. Craniotomies disrupt the normal parenchyma, activate immune cells/microglia, and show evidence of gliosis indicating mild injury to the brain. Behavioral data also supports that minor deficits can be detected from craniotomies alone [46].
Repetitive mild traumatic brain injury models are lacking, but the few models previously described give important insights into the association of chronic repetitive brain injuries and neurodegenerative diseases. It has been shown that repetitive mild traumatic brain injury increases cellular markers associated with Alzheimer’s disease [47]. DeFord and coworkers demonstrated one of the first animal models showing complex/spatial learning deficits following repeated mild TBI [48]. Uryu and colleagues have found that repetitive TBI accelerates deposition of amyloid-beta accumulation and oxidative stress, suggesting a synergistic relationship that can initiate the onset or drive further progression of Alzheimer’s disease [49]. Ojo and coworkers demonstrated in a mouse model that there was a significant increase in phosphorylayed-tau immunoreactivity in response to repetitive mild TBI, but not to a single mild TBI (mTBI). However, they did not find perivascular tau pathology, neuritic threads, or astrocytic tangles, all of which are typically seen in human cases of chronic traumatic encephalopathy [50]. Mouzon and coworkers demonstrated in a rodent model that single mTBI was associated with transient motor and cognitive deficits, but repetitive mTBI was associated with more significant deficits [51]. Meehan and colleagues found that the cognitive effects of multiple concussive events are cumulative when delivered in a particular period of “vulnerability,” and may in fact be permanent. However, as the interim period is prolonged between concussive injuries, significantly less adverse cognitive effects were seen over time [52]. Huang and coworkers also showed that mildly-impacted brain was more vulnerable to repetitive injury when delivered within three days following the initial TBI [53]. Mannix and coworkers found that repetitive mTBI produced long-term cognitive deficits independent of increased beta-amyloid or tau phopsphorylation, when TBIs were repetitively administered within a particular “vulnerability period.” Luo and colleagues reported on finding significant impairments in spatial learning and memory in the chronic period of repetitive mTBI, and also demonstrated robust astrogliosis, and immunoreactivity of phosphorylated tau protein in the brain on post-mortem pathological exams [54]. Perhaps one of the more ideal animal models for repetitive mTBI was presented by Kane and colleagues. This particular model differed from many of the existing models in that repetitive impacts were administered to animals that were actually unrestrained. Many current models entail securing the animal head in a type of fixation device, such as earbars. However, Kane’s model more closely paralleled human concussive injuries, as there was rapid acceleration of the free-moving head and body, a component that is key to the concussive element in human TBI [55].

Figure 4: Schematic diagram of a controlled cortical impact model of TBI. Brain trauma is produced by using a pneumatic impactor to impact exposed brain through a craniotomy.
The repetitive mTBI models described above involve different methods of inflicting a traumatic brain injury. The various models use both invasive and non-invasive techniques, with some models involving a variation of Marmarou’s weight drop technique, and others involving a craniotomy with a subsequent controlled cortical impact with a pneumatic piston, or fluid percussion [47-56]. Huang and coworkers have developed a rather interesting model that ultimately eliminates two of the largest confounding factors: performing a craniotomy, and using anesthetics. Both of these factors in and of themselves do have an impact on the neurological status of the animal, as the craniotomy has the potential to cause some degree of brain damage, while some of the anesthetics used are in fact neuroprotectants. Huang’s model entails using awake mice restrained in order to achieve head restriction. A helmet with a metal disc is used to cover the target area of injury (Figure 5). The injury device is a pneumatic controlled impactor similar to other CCI models [57]. The upside of this particular technique involves the elimination of two significant confounding factors as mentioned above.
The exact mechanism of the CTE remains unclear, but it likely results from some degree of progressive neuronal loss [58,59]. The typical history of an individual with CTE involves concussive and subconcussive blows over a period of time [60] .Other risk factors include presence of the ApoE3 or ApoE4 allele [61]. CTE caused by repetitive mTBI has been shown to progress many years after the inciting events have ceased.
There is some speculation that there is involvement of excitotoxic amino acids and abnormal microglial activation [60]. There is likely a cascade of events taking place leading to the pathological state known as chronic traumatic encephalopathy [58,59].
Pathologically, there is a patchy cortical distribution of neuralfibrillary tangles (NFT’s). This may suggest that the location of the NFT’s within the brain correlates with areas of direct mechanical trauma from blows to the head over a particular period of time. There may be a degree of ischemic injury taking place, as the observation has been made that tau deposition tends to be in the depths of cortical sulci [58]. Advanced stages of CTE are known to be associated with generalized atrophy of the brain, reduced brain weight, in addition to atrophy of the frontal and temporal cortices, and medial temporal lobes [58,62]. Grossly, there also appears to have ventriculomegaly of the third and lateral ventricles, as well as a cavum septum pellucidum and septal fenestrations [63,64].
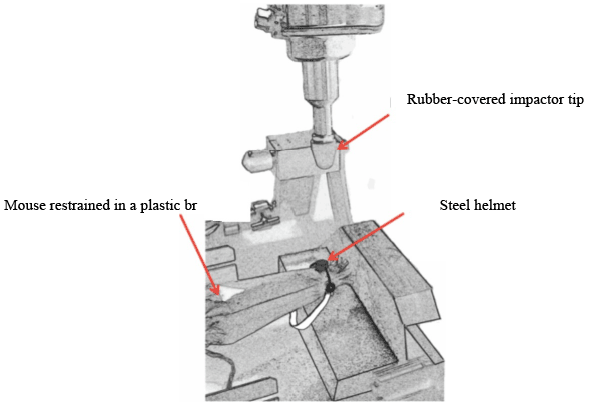
Figure 5: Schematic illumination of a chronic traumatic encephalopathy model of TBI. A non-anesthetized mouse is restrained in a plastic bag, and a steel helmet is placed on the head. Brain trauma is repeatedly delivered though impacting the helmet by the rubber-covered impactor tip using a modified controlled cortical impact device.
The blood brain barrier tends to break down following TBI. Local neurotoxins are released when this occurs, potentially explaining the perivascular clustering of NFTs [65]. Decreased microvasculature density and tortuosity has also been observed, corresponding to the laminar distribution of tau-positive NFT’s [66]. Tau does not appear to be the only inclusion culprit playing an important role in CTE. In animal models, the over expression of TDP-43 has been shown to cause neuronal degeneration and cell death [67]. The Boston University Neuroscience group led by Drs. Chin and Cantu found that other protein inclusions such as the DNA binding protein TDP-43 has been observed in brain specimens [62,68]. A common link between CTE and sporadic amyotrophic lateral sclerosis may also exist, as TDP-43 has also been observed in the brains and spinal cords of athletes presenting with both CTE and motor neuron disease [69]. McKee and colleagues observed pathological evidence that TDP-43 proteinopathy extended into the spinal cords of individuals with a history of repetitive TBIs. This may suggest that repetitive head injury not only causes cognitive and psychiatric disturbances, but also a pattern of motor neuron disease in some individuals afflicted with CTE [69].
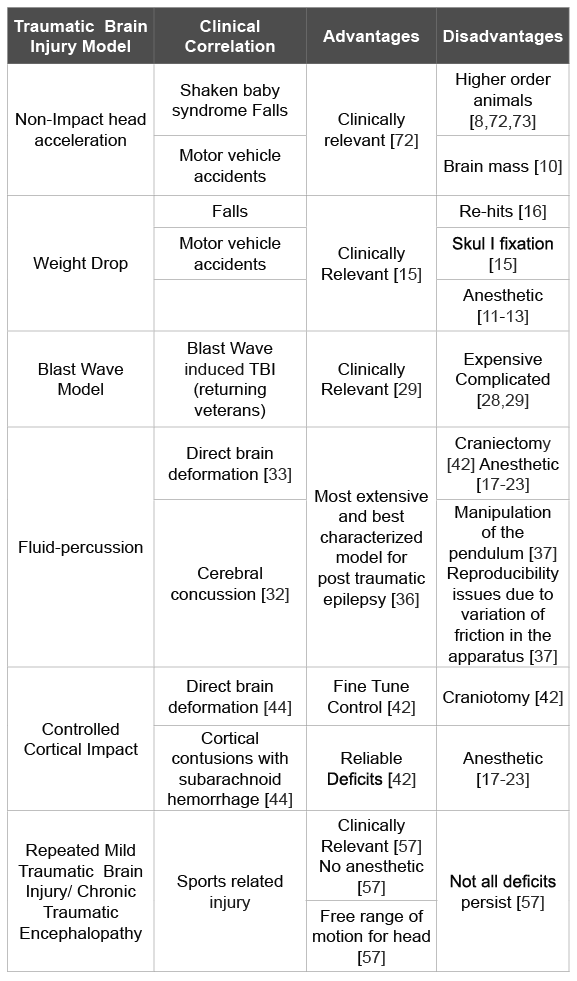
Table 1: A summary of the TBI models discussed in this article
Various closed head impact models exist. These have been designed in an effort to re-create the pathobiology and biomechanics of human concussive and diffuse brain injury [7]. Techniques discussed above such as CCI, fluid percussion, and weight drop, all utilize some degree of cortical impact resulting in a focal brain injury. Concussive events, however, involve a more global disturbance. Tornheim and colleagues developed a concussive cat model. The subjects sustained either a blow to the coronal suture or an oblique lateral impact with a Remington humane stunner [6,41]. This blunt trauma generated extra-axial hemorrhages, cerebral contusions, subarachnoid hemorrhages, as well as skull fractures [7,70]. This particular model has been utilized in a limited fashion due to the generation of hemorrhages and subsequent cerebral edema [70]. Goldman and coworkers created a rat model of moderate concussive injury. After the animal was anesthetized, a pendulum was designed to fall on the midline skull anterior to the coronal suture. Various gradations of impact could be applied by adjusting the applied force and impact angle. This particular model did not generate the same type of untowardly effects as the previous model discussed by Tornheim.
Goldman’s model did not result in any intra- or extra-axial brain hemorrhages, subarachnoid hemorrhage, cortical contusions, or skull fractures [7,71].
A realistic CTE model would entail some degree of repetitive mTBI delivered in a controlled, standardized fashion to animal subjects. Partial replication of the above mentioned model put forth by Goldman [71] would be highly considered, given its lack of significant complications and similarities to human concussive events. There would need to be a sham group, as well as a group receiving a single mTBI, in addition to the group of interest receiving repetitive mTBI over the course of a few weeks. It may be of interest to divide the repetitive mTBI groups into two separate groups: one group receiving repetitive mTBI on a routine basis in a close temporal relationship, and a second group receiving repetitive mTBI with a greater time interval between injuries. A free-moving head would also be highly considered as part of an ideal model, since acceleration is an etiological factor leading to concussive events in humans.
Of the few current models on CTE available, many of them fall short in that significant confounding factors such as anesthesia and performing craniotomies alter the neurological status of the subject. The vast majority of concussive events in humans involve active, awake, unrestrained, individuals, usually wearing some type of protective helmet. We should attempt to study this scenario to the best of our abilities in a scaled- down animal model.
Traumatic brain injury continues to be a significant social and medical issue plaguing over one million people every year. As we have outlined above, TBI comes in a variety of presentations, from severe life threatening diffuse cerebral edema, to minor alterations in consciousness. The particular subtype of TBI that is worrisome for individuals involved in sports continues to be repetitive mild brain injuries, with subsequent ensuing encephalopathy later in life, leading to significant neurobehavioral and psychiatric disturbances. There are a multitude of animal TBI models involving diffuse injuries, controlled cortical impact, as well as blast injuries (Table 1). However, a paucity of models exists culminating in the animal-equivalent of chronic traumatic encephalopathy in humans. The effort that needs to be made in future studies is to create an ideal CTE animal model for studying CTE pathogenesis and for developing therapeutic strategies for CTE.
This work was supported by the endowment of George W. Perkins, Jr.
Download Provisional PDF Here
Aritcle Type: Review Article
Citation: Galgano M, Russell T, Mc Gillis S, Toshkezi G, Chin L, et al. (2015) A Review of Traumatic Brain Injury Animal Models: Are We Lacking Adequate Models Replicating Chronic Traumatic Encephalopathy? J Neurol Neurobiol 2(1): doi http://dx.doi.org/10.16966/2379- 7150.117
Copyright: © 2015 Galgano M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access