Abbreviations
Aβ42: β-amyloid 42; Cdk5: Cyclin-dependent Kinase 5; DG: Dentate
Gyrus; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; NS: Sterile
Saline; HE: Hematoxylin and Eosin; NS: Sterile Saline
Background
AD is a progressive degenerative disease of the central nervous system
in the aged or aging period [1-3]. The main pathologies of AD, mostly
found in the hippocampus, cerebral cortex and subcortical tissue, are the
accumulation of amyloid plaques outside the cells [4], neurofibrillary
tangles in neurons [5], decreased synapses [6] and neuronal loss [7], etc.
Currently, the pathogenesis of AD has long been debated. Researchers
have proposed numerous hypotheses, including cholinergic injury [8],
Aβ neurotoxicity [9-14], hyperphosphorylation of tau [15-20], oxidative
stress [21,22] and the mechanism of metal which induces oxidative
neurotoxicity or amyloid aggregation [23,24]. Recent evidence suggests
that AD was not attributed to a single causative factor, but due to a variety
of concurrent effectors [25]. Furthermore, the combined effects of Aβ and
tau might serve as a major etiological factor.
In the development of Aβ neurotoxicity, it was found that soluble
oligomers of Aβ (i.e., amyloid beta derived diffusible ligands (ADDLs),
nonfibrillar ligands derived from Aβ42) induced the strongest toxicity
[26-29]. The presence of ADDLs leads to hyperphosphorylation of
tau, and hyperphosphorylated tau then dissociates from the neural
microtubules. As a result of the increased number of free tau, the
stability of the cytoskeleton is disrupted. The abnormal change in the
cytoskeleton generates large amounts of neurofibrillary tangles which are
the other principal pathological change of AD. Moreover, Aβ is believed
to catalyze hyperphosphorylation of tau through some intermediates like
phosphorylated kinases [30, 31].
Tau, a microtubule-associated protein, mainly in axons, is located in the
cell body in the early development of neurons, and then moves toward the
axon after the maturation of neurons. Also, tau has the effect of stabilizing
neurons. The association of neurofibrillary tangles (NFT) which is a major
pathological feature of AD with tau is significant [32]. Recent clinical
testing revealed that the total amount of tau and the content of p-tau in
AD patients’ blood and cerebrospinal fluid were substantially higher than
those in patients with normal and non-neurological groups [33].
In addition, cyclin-dependent kinase 5 (Cdk5) can inhibit microtubule
binding to tau which triggers tau hyperphosphorylation. Recent evidence
has also confirmed that primary cultured hippocampal neurons coincubated
with Aβ stimulate tau phosphorylation and cause unusual
activation of Cdk5, altering the distribution of tau and the levels of
phosphorylated tau in neurons [34].
Above all, owing to the complicated pathogenesis of AD, this study
will emphasize the effect of Aβ to tau phosphorylation. Researchers
have revealed that Aβ plays a significant role in tau phosphorylation in
vitro; however, it didn’t directly describe in vivo. In this study, our data
suggest that ADDL treatment induces hyperphosphorylation of tau via an
increase of Cdk5.
Methods
Aβ preparation
Aβ42 (soluble Aβ oligomers) were purchased from AnaSpec (San Jose,
CA). 500 μg Aβ42 was dissolved in 100 μl 1% NH4 OH to make a 5 μg/μl
stock solution, which was placed in aliquots at -20°C. After adding NS, a
working solution was diluted to a concentration of 2 μg/μl and incubated
at 4°C for 24 hours, obtaining the oligomeric species of Aβ.
Aβ intrahippocampal injection
One hundred nine-week-old male BALB/c-wild type mice (weight 20-
28 g) were obtained from the Experimental Animal Center of Guangzhou
University of Traditional Chinese Medicine. All animal procedures
performed were approved by the Animal Ethics Review Committee and
confirmed the guidelines for the Care and Use of Laboratory animals of
Guangdong Pharmaceutical University. These mice should be bred in a
Specific Pathogen Free (SPF) environment of separate ventilated cages.
The mice were randomly assigned to a normal group (n=20), a NSinjected
group (n=40) and an Aβ42-injected group (n=40). For stereotactic
injection, mice were anesthetized with 0.4% sodium pentobarbital
in a dosage of 0.2 mL/10 g. All mice in the Aβ42-injected group were
injected was into the CA1 area in the hippocampus bilaterally with 2 μl
Aβ42 peptide (2 μg/μl) while mice in the NS-injected received 2 μl NS,
and mice in the normal group did not receive any surgery or treatment.
The coordinates from bregma used for the CA1 region were as follows:
anteroposterior, -2.3 mm; mediolateral, ±1.8 mm; dorsoventral, -2.0 mm.
The injection was performed by using a 25 ml Hamilton syringe driven
by a minipump (KDS Model 310 Plus, KD Scientific, Holliston, MA) at
0.8 μl/min. After injection, the needle was kept in the area for another
2 min, and then slowly retreated to prevent spill. Following 7, 14 and 21
days post-injection, the mice was anesthetized and brains were dissected
immediately. The right hemisphere was fixed in 4% paraformaldehyde for
48 hours for histological detection. The left hippocampus was snap-frozen
in a pre-cooling vial in liquid nitrogen and finally stored at -80°C for
western blot analysis and transcription polymerase chain reaction (RTPCR)
testing.
Histological staining
Paraffin-embedded tissue was sectioned on microtome and coronal
slices with a thickness of 6 μm were collected. Serial sections (six sections
per hippocampus) at an interval of 60 μm between -1.8 mm and -3.2
mm from the bregma were chosen. Mounted sections were washed
with distilled water and stained with alum hematoxylin followed by
differentiating with 0.3% acid alcohol. Then rinsed in distilled water,
sections were stained with eosin for 2 min and dehydrated for mounting.
Immunohistochemical staining
After dewaxing with water, sections were incubated with 3% H2 O2 at room temperature (RT) for 15 min, placed in a blocking solution (10% BSA in 0.3% Triton X-100/0.1M PBS pH 7.4) at RT for 30 min, and then binding to the unlabeled primary antibodies at 4°C overnight occurred. The following primary antibodies and working condition were used: rabbit polyclonal anti-Aβ42 (ab10148, Abcam, Cambridge, UK, 1:250), rabbit polyclonal anti-Cdk5 (ab151233, Abcam, Cambridge, UK, 1:100) for immunofluorescence, and rabbit polyclonal anti-p-tau (ab10891,
Abcam, Cambridge, UK, 1:300) for immunofluorescence. On the second
day, after washing three times, sections were incubated with secondary
antibodies: biotinylated goat anti-rabbit (ab128978, Abcam, Cambridge,
UK, 1:1000) at RT for 1 hour. Negative controls were incubated without
primary antibody. Immunostaining was achieved by using the avidinbiotin
complex (Vectastain: Vector Laboratories, Burlingame, CA) and
diaminobenzidine plus Nickel (DAB Kit: Vector Laboratories) as the
chromogen.
For density of Aβ42, Cdk5 and p-tau, randomly selected areas within
the hippocampal CA1 and DG regions, were imaged using Olympus BX51
light microscopy (Olympus, Tokyo, Japan).
Western boltting analysis
Protein extraction from the hippocampus was separated by RIPA Lysis
Buffer (Beyotime, Shanghai, China) (50 mM Tris (pH 7.4), 150 mM NaCl,
1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, protease inhibitor
cocktail (sodium orthovanadate, sodium fluoride, EDTA, leupeptin),
and 1 mM phenylmethylsulfonyl fluoride). Quantitative analysis of
the protein was determined with a BCA protein assay kit (Beyotime,
Shanghai, China). The protein samples were separated on 12% SDS-PAGE
gels, and electronically transferred to a PVDF membrane at 0.8 mA/cm2
for 1.5 hours. Then, the membrane was rinsed with 5% nonfat dry milk
in 20 mM Tris-HCl (pH 7.4) with 0.05% Tween20(TBS-T) for 1 hour at
RT, and incubated with primary antibodies: rabbit polyclonal to anti-ptau(sc-101813,
Santa Cruz Biotechnology, 1:500), rabbit polyclonal antiCdk5
(sc-173, Santa Cruz Biotechnology, 1:500) and mouse monoclonal
anti β-Actin (Santa Cruz Biotechnology, 1: 1000) at 4°C overnight. After
being washed with TBS-T, the membrane was incubated with HRPconjugated
IgG (13688-1-AP, Proteintech, Chicago, IL, 1:1000) for 1 hour
at RT, and washed with TBS-T again. Blots developed with enhanced
chemiluminescence (ECL, Thermo Scientific, Waltham, MA, USA).
Analysis of densitometric quantitation was performed with Image J
Software (NIH, Bethesda, MD, USA) and standardized with β-Actin. The
above samples were run in triplicate.
RT-PCR analysis
Tissue extracts were harvested and rinsed with phosphate-buffered
saline (PBS) at corresponding time points and total RNA in the treated
sections was extracted according to the total RNA extracting kit (AM1830
, Life Technologies). A solution was added consisting of 2 μL (5 mmol/μL)
dNTP, 1 μL oligo(dT), 1 μL (200 U/μL) reverse transcriptase (m-mulv),
pH 8.3 RT buffer (250 mmol/L Tris-HCl, 250 mmol/L KCl, 20 mmol/L
MgCl2, 50 mmol DTT) and deionized water. The total sample volume was
20 μL. Samples were incubated at 42°C for 1 hour and the reaction was
stopped by heating at 70°C for 10 min. Reverse transcriptase was used
to synthesize the first strand cDNA from an equal amount of the RNA
sample following the manufacturer’s instructions. About 40 cycles of the
PCR reaction were used to cover the linear range of the PCR amplification.
The band densities were scanned with a densitometer (Bio-Rad, United
States).
Statistical analysis
Results were analyzed with SPSS 16.0 software (IBM, Armork, NY,
USA), with significance at P <0.05. Quantitative data were documented
as the mean ± SD. Fisher’s exact tests were performed on categorical data
to verify the difference of degenerating neurons, Aβ aggregation, tau
hyperphosphorylation and Cdk5 expressions between Aβ42-injected and
NS-injected group, respectively. When comparing Aβ42-injected and NSinjected
group, a two-tailed t-test was conducted for protein levels of tau
and Cdk5 in the hippocampus. The Kruskal-wallis one-way test was used
to test tau mRNA level among to the normal group, the NS-injected group
and the Aβ42-injected group.
Results
Aβ42-injection caused neuronal degeneration
To determine whether Aβ42 are sufficient to trigger hippocampal
neurodegeneration, we analyzed histological alternations of morphological
structure using HE staining after treatment with Aβ42 after 7 days. In the
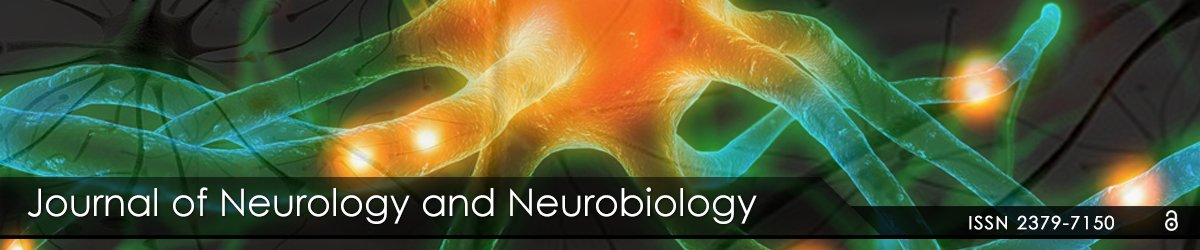
normal group (Figure 1A), HE staining showed that the neurons were in
alignment (Figure 1B). In the sterile saline (NS)-injected group (Figure
1C), the surgery-induced injury was limited to the zone of the needle track
(Figure 1D). However, neurodegeneration was significantly shown in the
area where Aβ42 was injected (Figure 1E). In addition, Aβ42-injected
animals have degenerating neurons in the Cornu Ammonis area 2 (CA2)
(Figure 1F). This result demonstrated that Aβ42 triggered neurotoxicity of
neurodegeneration in hippocampal pyramidal neurons.
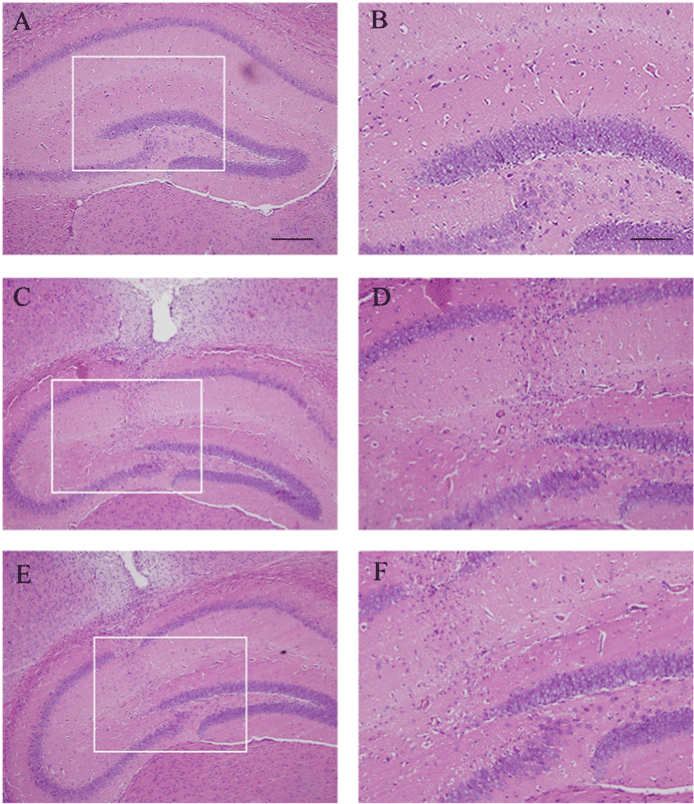
Figure 1: Histological changes in the hippocampus. (A) HE staining in normal hippocampus of BALB/c mice. (C) HE staining in NS-injected hippocampus. (E) HE staining in Aβ42-injected hippocampus. (B, D, F), respectively, represent the region of high magnification in (A, C, E). (F) HE staining showed neuronal loss at the injection site surrounding Aβ peptide. Scale bar=200 μm in (A, C, E) and 50 μm in (B, D, F).
Deposition of soluble Aβ oligomers in the hippocampus
After injecting Aβ42 into the hippocampus, Aβ42 were deposited and
metabolized. The level of Aβ42 in the hippocampus was measured using
anti-Aβ antibody. When observing the expression of Aβ for 7 days, 14
days and 21 days, we found that Aβ accumulation was much higher on
day 7 than on the other days (data not shown). Therefore, the mice after
Aβ injection for 7 days were selected. In the NS-injected group (Figure
2A), Aβ expression was barely observed at the injection site (Figure 2B).
In contrast, Aβ expression was significantly expressed in SGZ of dentate
gyrus (DG), which showed in approximately 94% of 40 Aβ-injected
animals (Figures 2C and 2D) versus none of 40 NS-injected animals
(P<0.0001, Fisher’s exact test).
Aβ42-injection induced tau hyperphosphorylation
Our results above showed that Aβ42 primarily deposited in DG.
We asked whether injecting Aβ42 into the hippocampus enhanced tau
phosphorylation. We assessed the level of phosphorylated tau after
injected with Aβ42 (Figure 3D) using immunofluorescence. Compared
to NS-injection (Figures 3A-3C), tau hyperphosphorylation was observed
in 35 of the 40 Aβ42-injected animals in both CA1 (Figure 3E) and DG
(Figure 3F) versus 8 of the 40 NS-injected animals (P<0.0001, Fisher’s
exact test). Thus, Aβ42 accelerated hyperphosphorylation of tau.
Aβ42-injection activated Cdk5 when inducing tau hyperphosphorylation
Cdk5, a member of the cyclin-dependent protein kinase (CDK) family, can lead to neurological dysfunction and trigger neurodegenerative diseases when it is abnormal. Also, increased Cdk5 may hasten tau
phosphorylation [35]. We tested Cdk5 expression in primary cortical
neurons within Aβ42-injected within Aβ42-injected and NS-injected
groups in the hippocampus by immunofluorescence (Figure 4). We
observed Cdk5 in 32 of 40 (P<0.05, Fisher’s exact test) Aβ42-injected
animals (Figure 4D) both in the CA1 (Figure 4E) and DG (Figure 4F),
respectively, versus 8 of 40 NS-injected animals (Figures 4A-4C, P <0.0001,
Fisher’s exact test). In previous studies, they indicated that Cdk5 kinase
activity in phosphorylating tau dramatically increased in the presence of
p25 compared to p35 in vitro [36]. And the enhancement of Cdk5/p25
triggered tau hyperphosphorylation [37]. It can be speculated that Aβ42
may promote tau phosphorylation by activating Cdk5, which results in
hyperphosphorylation of tau in AD pathology.
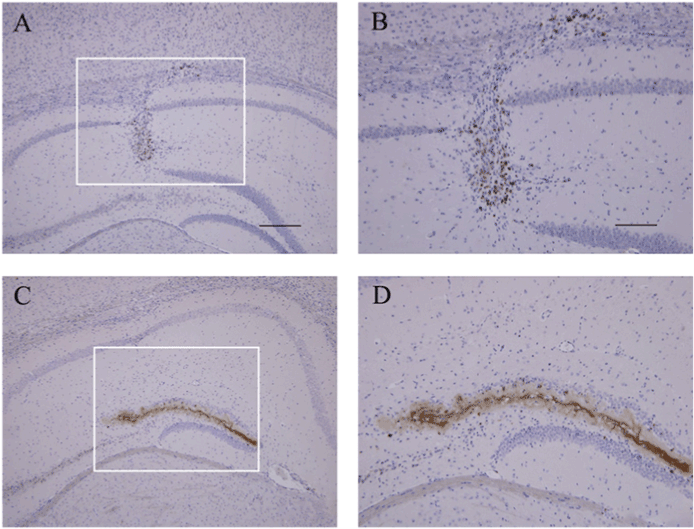
Figure 2: Immunostaining of Aβ in the hippocampus. (A) NS-injected group. (B, D), respectively, represent the region of high magnification in (A, C). (B) Higher magnification of negative Aβ aggregation at the injection site from NS-injected hippocampus. (C) Aβ42-injected group. (D) Aβ aggregation in SGZ of DG from Aβ42-injected hippocampus. Scale bar=200 μm in (A, C) and 100 μm in (B, D).
Figure 3: Immunofluorescence for expression of hyperphosphorylated tau in hippocampus. (A) Tau staining in control group (BALB / c + NS). (B, C) represent the region of high magnification in (A). (B) Negative au expression located in CA1. (C) Negative tau expression located in DG. (D) Tau staining in Aβ42-injected group (BALB / c + Aβ42). (E, F), respectively, represent the region of high magnification in (D). (E) p-tau located in CA1. (F) p-tau located in DG. Scale bar=100 μm in (A, D) and 50 μm in (B, C, E, F).
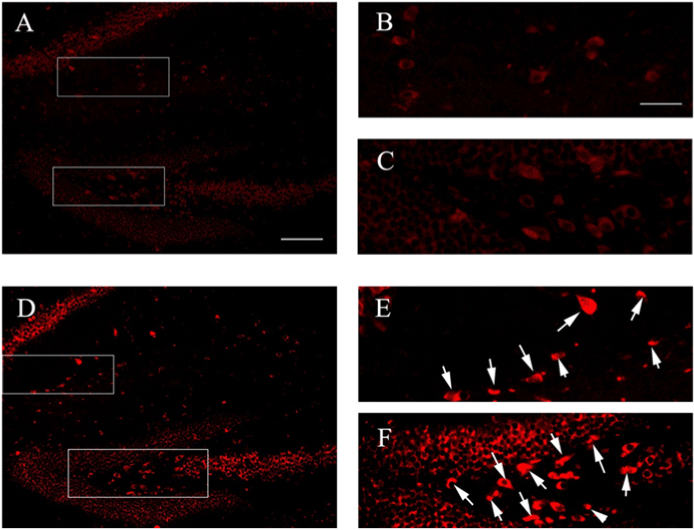
Figure 4: Immunofluorescence for expression of Cdk5 in the hippocampus. (A) Cdk5 staining in control group (BALB / c + NS). (B, C) represent the region of high magnification in (A). (B) Negative Cdk5 expression located in CA1. (C) Negative Cdk5 expression located in DG. (D) Cdk5 staining in Aβ42-injected group (BALB / c + Aβ42). (E, F), respectively, represent the region of high magnification in (D). (E) Cdk5 located in CA1. (F) Cdk5 located in DG. Scale bar=100 μm in (A, D) and 50 μm in (B, C, E, F).
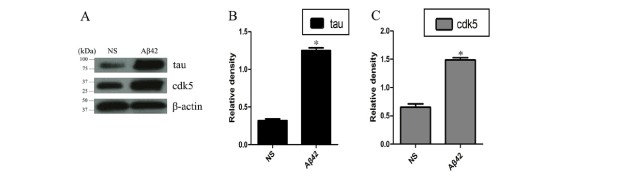
Figure 5: Protein levels of tau and Cdk5 in hippocampus 7 days after
Aβ42 injection. (A) Detection of protein levels of tau and Cdk5 using western blot. (B) Histograms represent the relative density of tau. Asterisks indicate data significantly different from those of NS injection (*P <0.01, student two-tailed t test). n=10 mice per group; error bars, SEM. (C) Histograms represent the relative density of Cdk5. Asterisks indicate data significantly different from those of NS injection (*P <0.01, student two-tailed t test). n=10 mice per group; error bars, SEM.
Aβ42-injection in hippocampus showed increase of Cdk5 and hyperphosphorylation of tau
To further confirm the mechanism of whether Aβ may activate the
enzyme of phosphorylated tau-Cdk5, which led to hyperphosphorylation
of tau, we assessed both the phosphorylation state of tau proteins (p-tau)
and the level of Cdk5 in the hippocampus, which were assayed by
quantitative western blotting with epitope-specific antibodies after being
cultured for 7 days (Figure 5A). The analysis of gray value in western
blot displayed a ~3.9-fold higher in p-tau protein in the hippocampus
7 days after Aβ42-injection (Figure 5B), indicating that an increased
p-tau level might be related to Aβ42-induced neurodegeneration.
Meanwhile, we found ~2.3-fold difference of Cdk5 enzyme between the
Aβ42-injection and NS-injection (Figure 5C). Thus, Cdk5 catalyzed tau
hyperphosphorylation, consistent with a positive role in AD.
Total protein tau in Q-RT-PCR validation
Hyperphosphorylation of tau in neurons results in tau dissociating from
neural microtubules and becoming free tau, which increases the amount
of total protein tau in neurons. To test and verify whether total value of
tau was regulated by the Aβ oligomeric, the melt and amplification plots
were carried out via Q-RT-PCR. Tau mRNA 7 days after Aβ42 injection
was significantly higher than that of the control and saline-injected groups
(P<0.05, Kruskal-Wallis test).
The delta cycle threshold (CT) value of tau from Aβ42-injection was
10.32, while that of the control and saline-injected groups was about 11
(Table 1, P<0.05, Kruskal-Wallis test). The results of relative quantity
among three groups, suggesting that expression of tau 7 days after Aβ42
injection significantly increased, while the others were not statistically
different (P<0.05, Kruskal-Wallis test).
Discussion
Finding the specific relationship between Aβ and tau, in this study,
further enriches AD etiology. Here, using synthetic oligomer Aβ42 injected
into mouse hippocampi, we observed this single factor was sufficient
to cause neurodegeneration in the hippocampal cortex and induce
hyperphosphorylation of tau in the CA1 and SGZ of DG. Meanwhile, we
showed that Cdk5 was activated by Aβ42, suggesting that this new target
spot might intervene in the occurrence of AD.
| Sample |
CT value
of Tau |
CT value
of GAPDH |
ΔCT |
ΔΔCT |
Relative
Quantity (2-ΔΔCT) |
| Control |
16.91 |
6.14 |
10.77 |
0 |
1 |
| NS-injection |
16.99 |
6.15 |
10.84 |
0.07 |
0.952637998 |
Aβ42-
injection |
20.37 |
6.05 |
10.84 |
3.45 |
0.091505355* |
*P < 0.05 compared to control group, Kruskal-Wallis test
Table 1: The cycle threshold values (CT) of real-time Q-RT-PCR
Given that Aβ promoting tau hyperphosphorylation has long been
proposed, one is unable to specify which direct role of Aβ is responsible
for this process, as free-state, oligomeric peptides or amyloid plaques.
Moreover, some studies have performed that oligomeric Aβ has a higher
toxicity and is an important factor in the process of AD [23,24,38,39].
Also, any AD animal models show cognitive impairment, especially
AD transgenic mice, those with different genetic backgrounds, mutable
sites of tau, and a variety of different features of amyloid deposition or
abnormal behaviors, are likely to lead to inconsistencies in neurogenesis.
For example, some study found out the expression of Aβ42 was aggravated
in the brain tissues of neonatal AD transgenic mice using sevoflurane
anesthesia compared to neonatal naive mice. Therefore, our study selected
BALB / c-wild type male mice as experimental subjects [40]. After an
injection of Aβ42 into mice, tau moves toward the dendrites, resulting in
some changes in the physiological characteristics of neurons. Tau in the
dendrites of neurons can inhibit the transport of organelles, reduce the
supply of ATP, and induce synaptic joint recession. Compared to normal
neurons, the processus spinosus of neurons injected with Aβ42 decreased
by approximately 75%. In the meantime, owing to the redistribution
of tau, the number of neural microtubules also declines significantly.
Therefore, Aβ42 affect neuronal function by inducing the redistribution
of tau in dendrites, which induces neuronal apoptosis [41,42].
Furthermore, under normal circumstances, only 2-3 phosphate
groups are in modification of tau, so tau can keep a balance between
phosphorylation and dephosphorylation, which regulates the
cytoskeleton. Conversely, linking to 9-10 phosphate groups, the coexpression
of Aβ42 and tau leads to enhanced hyperphosphorylation of
tau, and then the aggregation of tau leads to the formation of neurotrophic
factors (NFTs) in pathological situations. Owing to loss of its biological
function, hyperphosphorylated tau is insufficient to promote microtubule
accumulation and maintain a stable cytoskeleton. After dissociating
from the microtubules, hyperphosphorylated tau competitively binds
to the normal tau, resulting in increased free tau but reduced normal
physiological functioning of tau [43,44]. It was confirmed that this form
of Aβ induced tau hyperphosphorylation in hippocampal neurons, and
then caused rupture of the microtubule cytoskeleton as well as neuron
degeneration [45]. Therefore, we hypothesized that soluble oligomeric
forms of Aβ may play the principal role in triggering cytoskeletal changes
as well as neuronal toxicity
Our observation on the expression of Aβ42 effects on tau
hyperphosphorylation has relevance to increasing activity of Cdk5.
To our knowledge, the presence of non-cyclin protein p35 within the
cerebral cortex is a Cdk5 activator, but the limitations of its distribution
restrict activation of Cdk5 [35]. When subjected to Aβ42 stimulation, p35
is cleaved into p25 and the other fragment toward calpain activity. p25,
whose half-life is 5-10 times longer than p35, completely activates Cdk5.
p25-Cdk5 complex lacks the membrane-anchoring signal motif after
cleavage, leading to its change of position within the cell [46-48]. On one
hand, Cdk5 can directly trigger phosphorylation of tau protein [49], but
on the other, Cdk5 can play a role in tau by regulating the relevant kinase
or phosphatase [50]. Therefore, Cdk5 has a large effect on abnormal
phosphorylation of tau related to the occurrence and development of AD.
We still found several limitations in this study. The primary limitation
was the inability to prove whether Aβ triggered p-tau or p-tau triggered
Aβ through the reduction to absurdity. We could not evaluate the change
of Cdk5 or p-tau when it was lack of Aβ. Further limitation was different
behavior types of analysis; however, the data was obtained prospectively.
Taken together, our study confirms that Cdk5 is involved in the disease
process of neurodegenerative diseases, whose long overexpression can
result in cell death. Aβ induced Cdk5 activity increased by p25, which
further triggers the hyperphosphorylation of tau, leading to neuronal
degeneration and cell death [51,52]. Recent studies have proven the
developmental and metabolic disorder of Aβ as well as accelerated tau
hyperphosphorylation, in addition to significantly increasing expression
level of Cdk5, in the pathogenesis of AD [53]. Rather, such associations
among the above three are firmly suggestive.
Conclusions
In this study, we demonstrated that Aβ, a source of AD, whose
metabolic abnormalities lead to excessive accumulation of toxic soluble
oligomeric forms of Aβ in the brain, is likely to contribute to the profound
influence in pathological features of AD, by raising Cdk5 expression and
tau hyperphosphorylation.
Acknowledgments
We thank Prof Dr W. Qing Mei, Department of Physical Medicine and
Rehabilitation, Spaulding Rehabilitation Hospital, Boston, MA, USA, for
advice regarding hyperphosphorylation of tau and total tau in neurons
and Yuxin Ma and Jing Liu for excellent technical support.
Authors’ contributions
Ms. Shanshan Li takes charge in the preparation of animal models and
the experiments of neuronal histochemistry. Ms. Sumin Tian is charge of
western blot and PCR. Ms. Lingzhi Sun assists Ms. Shanshan Li and Ms.
Sumin Tian in all the experiments. Mr. Zhihao Liang, Ms. Xiaohui Cheng
and Mr. Han Wang give assistance for preparation of animal models and
postopreative care for animals, Ms.Yuxin Ma and Dr. Jing Liu provide
guidance on immunohischemistry, fluorescence chemistry and etc. Dr.
Guoying Li is responsible for the design and implementation of this
subject. Dr. Qing Mei Wang gives advice to the design.
Competing Interests
All authors declare that they have no competing interests.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No.30840073).