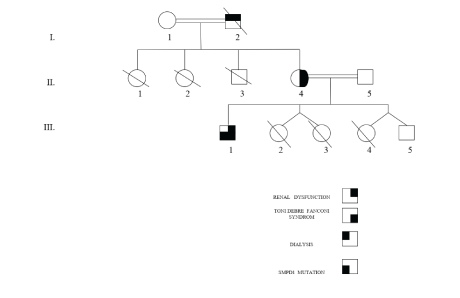
Figure 1: Family pedigree of the Algerian family with clinical Toni Debré Fonconi syndrome (I 2, II 4 and III 1) associated with Niemann Pick B syndrome (III 1).
Ghalia Khellaf1* M Saidani1 S Missoum2 T Rayane3 L Kaci4 M-C Gubler5 M Benabadji1
1Benkheda Youssef University, Department of Nephrology, Beni Messous Hospital, Algeria*Corresponding author: Ghalia Khellaf, Benkheda Youssef University, Department of Nephrology, Beni Messous Hospital, Algeria, E-mail: g.khellaf@yahoo.fr
Background: Toni Debré Fanconi syndrome is characterized by a generalized dysfunction of the proximal tube. The isolated, familial syndrome is rare. It is transmitted according to the autosomal dominant mode. Niemann-Pick disease (NP) is an equally rare autosomal recessive disorder characterized by lysosomal accumulation of sphingomyelin in body cells. The disease is caused by mutations in the SMPD1 gene that cause lysosomal acid sphingomyelinase deficiency.
Case Report: We report a case of a 28-year-old patient from first-degree consanguineous marriage, family history of polydipsia and polyuria, staff hospitalized for moderate renal impairment and asthenia, the finding of tubular proteinuria associated with hypouricemia, hyperphosphaturia and hypercalciuria, leads to the diagnosis of Toni Debré Fanconi syndrome. On the other hand, the particular face of the patient, the existence of hepatosplenomegaly, hypertriglyceridemia and thrombocytopenia lead to the diagnosis of Niemann Pick type B syndrome, diagnosis confirmed by the biochemical assay and the genetic study.
Toni Debré Fanconi syndrome; Niemann Pick B syndrome; Renal failure
Toni Debré Fanconi syndrome is a complex tubulopathy characterized by a generalized dysfunction of the proximal tube [1,2] causing urinary leakage of components of glomerular filtrate, amino acids, glucose, phosphates, bicarbonates, calcium, potassium, uric acid and other solutes. This syndrome may be secondary to intoxication (mercury, lead), an inherited metabolic disease (cystinuria, galactosemia), and cancer (myeloma). Otherwise, the Fanconi syndrome is said to be primitive; it is then sometimes hereditary and transmitted according to the autosomal dominant mode [1,2]. The symptoms of Fanconi syndrome are the result of renal leakage: dehydration, metabolic acidosis, hypocalcemia, responsible for bone fragility, rickets and growth retardation in children, or osteomalacia in adults. Treatment of the cause, when identified and curable, causes healing. In other cases, the treatment is that of the symptoms: rehydration, calcium intake, Vitamin D, alkaline, etc. Autosomal recessive inheritance, Niemann-Pick disease is due to acid sphingomyelinase deficiency [3]. We distinguish type A, type B, and intermediate forms, called A/B. Type A is manifested in the first year of life by digestive disorders, an alteration of the general state, a major hepato splenomegaly, sometimes brownish skin spots or xanthomas, then a neurological involvement with hypotonia between 6 and 12 months, stopping psychomotor development, spasticity with are flexia. Neurological degradation and frequent pulmonary infections lead to death around the age of 3 years. Type B forms are less severe: the neurological involvement is absent and the age of onset is very variable, until adulthood. The most consistent sign is hepatosplenomegaly, with occasional recurrent pulmonary infections, interstitial lung disease, joint pain, diarrhea, stunting and puberty [3]. Intermediate forms A/B are described with moderate, slow or late neurological involvement. The diagnosis is confirming by the determination of the activity of acid sphingomyelinase, the residual activity of which does not make it possible to differentiate the types. A prenatal diagnosis is possible. Recombinant sphingomyelinase treatment is on evaluation. Type C NP, originally described with types A and B, has a different origin; it is linked to mutations of the NPC1 or NPC2 genes leading to a lack of esterification and intracellular transport of cholesterol [4].
We report a case of a patient (Figure 1), 28 years old hospitalized for exploration of moderate renal insufficiency, unlabeled discovered during low back pain and asthenia occurred at the end of the Ramadhan period. Among his personal antecedents, there is prematurity at 8 months, polyuria-polydipsia since childhood and hepato-splenomegaly accidentally discovered during pediatric hospitalization at the age of 4 years. The etiological assessment had not led to any conclusion. At the age of 17, the patient develops vulgaris acne lesions and nasal turbinate hypertrophy, associated with palpebral oedema, without oedema of the lower limbs. On examination, at the age of 28, the development of weight, height and puberty is normal with a size of 172 cm/70 kg. The face is particular; associating a hypertrophy of the turbinat’s nose, an eruption in wings of butterfly and a palpebral oedema. Macules and papules are present in the thorax and back. The abdomen is supple, with a huge hepatosplenomegaly. The BP is normal at 120/80 mmHg. The rest of the clinic exam with no abnormalities. The biochemical assessment confirms the moderate renal insufficiency: creatinemia to 21 mg/l with clearance to 47 ml/mn, normal uraemia to 0.31 g/l. The level of uricemia is low: 20 mg/l. Blood ionogram and blood glucose (0.94 g/l) are normal. The diuresis is 3l/24 hours, at the urinary strip the pH is at five, there is proteinuria and glycosuria. The assessment confirms proteinuria at 1.24 g/24 h, associated with hyperuricuria at 780 mg/24 h, hypernatriuria at 200 meq/l, hyperkaliuria at 31 meq/l, hypercalciuria at 435 mg/24 h, hyperphosphaturia at 1117 mg/24 h, has neither hematuria nor leucocyturia.
Figure 1: Family pedigree of the Algerian family with clinical Toni Debré Fonconi syndrome (I 2, II 4 and III 1) associated with Niemann Pick B syndrome (III 1).
On the radio, the kidneys are slightly diminished in size (right kidney: 9 cm, left kidney: 9.2 cm), they are discreetly hyperechoic but keeping a good corticomedullary differentiation. Renal biopsy involving the cortico-medullary region allowed examination of 6 to 15 glomeruli (Figure 2). Two are transformed into sealing loaves, the others are morphologically normal, without conventional immune fluorescence deposits. The vessels are normal. Two small foci of nonspecific interstitial fibrosis are present, one of which surrounds some atrophic proximal convoluted tubule (Figures 3 and 4). In total, all the biological results lead to the conclusion of the diagnosis of Toni Debré Fanconi syndrome, probably genetic given the family history of polyuria. Indeed, the maternal grandfather (Pt I 2) polyuro-polydipsic since childhood and had developed a bilateral renal lithiasis from the age of 35, treated by extracorporeal lithotripsy. In hemodialysis since the age of 65, he died at the age of 72. The mother (Pt II.4) patient’s 52-year-old, who had been consanguineous, had nycturia until she was 14 years old. The diuresis estimated in childhood was 3 to 4 litres/24 hours. She has also developed bilateral renal lithiasis treated repeatedly with lithotripsy. She complains of lumbar pain and asthenia. Four pregnancies, two of which were followed by an abortion, the death on the day of their birth of 3 premature infants (6 and 8 months) and the birth, after 8 months, of 2 boys, including the patient, low birth weight (2 kg 700 g and 2 kg 500 g). During a consultation in nephrology in the dialysis center of the patient (I 2), the clinical examination and the radiological assessment are without particularities, the TA is correct at 120/70 mmHg. There is a moderate renal insufficiency: clearance of the creatinine at 65 ml/min, uremia at 0.24 g/l, associated with hypouricemia at 10.34 mg/l. The blood glucose (1.02 g/l), serum calcium (88 mg/l) and phosphoremia (30 mg/l) are within normal limits, as well as the blood ionogram, the blood count formula, the lipid profile, the protidemia and albuminemia. The urinary assessment shows minimal proteinuria at 400 mg/24 h, glycosuria at 3+, hyperuricuria at 1215 mg/24 h, hypernatriuria at 292.5 meq/24 h, and hyperkaliuria at 105 Meq/24 h. But further investigations are necessary after the presence of chronic hepatosplenomegaly. The liver test is normal and the viral serology, HIV, hepatitis B and C, is negative. Cholesterolemia is normal at 1.65 g/l but there is hypertriglyceridemia at 5.31g/l. The immunoassay provides no evidence for lupus. The hematological assessment shows thrombocytopenia at 92000/mm3 without anemia or leukopenia. The blood smear is normal and bone marrow biopsy reveals the presence of large foam cells suggestive of metabolic overload disease, particularly in this adult, Gaucher disease or Niemann Pick. The diagnosis of Niemann Pick disease is confirmed by the results of biochemical and genetic studies: the presence of a very low level of 52 pmol/spot 20 h (norms 200-3500) of acid sphingomyelinase and demonstrated in the SMPD1 gene of a homozygous deletion C. (1829_1831delGCC) leading to the loss of arginine 608 (deltaR608) of the protein. In Niemann Pick’s disease, chest X-rays reveal diffuse interstitial pulmonary lesions. Echocardiography is normal. Abdominal ultrasound shows the heterogeneous nature of hepatomegaly, related to the presence of multiple echogenic nodular formations, the homogeneous appearance of the spleen, and the presence of a hypoechoic nodule of the right adrenal lodge. Doppler regains chronic hepatomegaly with signs of portal hypertension on intrahepatic block, with no signs of decompensation.
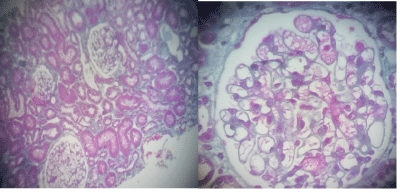
Figure 2: Renal biopsy of the patient (III 1) Optical microscopy with Trichrome Masson stain. Optically normal glomeruli.
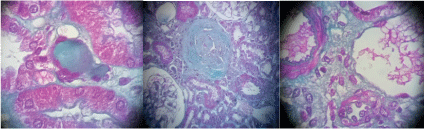
Figure 3: Renal biopsy of the patient (III 1). Optical microscopy with Trichrome Masson stain. Non-specific tubulo-interstitial lesions with 02 fibrosis centers, one of which contains some atrophic proximal convoluted tubules.
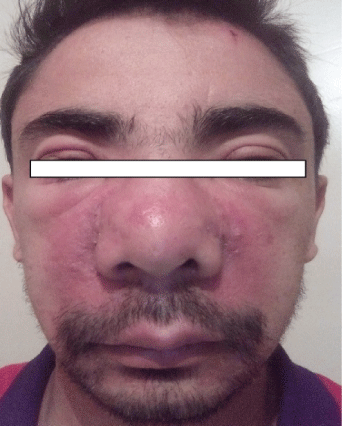
Figure 4: Facies of the patient (III 1). Eruption of papules in both nasolabial folds associated with enlarged nasal turbinates and palpebral edema.
The radiological examination confirms the hypertrophy of the cornets, without deviation of the nasal septum; the sinuses are free. The fundus and the examination with the slit lamp show no particularity. The biopsy of the salivary glands is normal. Revision of the renal biopsy, after staining by May-Grünwald Giemsa, oriented by the presence of “foam cells” in the marrow, does not find any particular deposits in the renal cells.
The association of two genetic pathologies, one autosomal dominant and the other recessive, is quite exceptional. It can be at the origin of diagnostic wandering, one of the diseases masking the other. In the case of our patient, with moderate chronic renal failure, family history of polyuria, and results of blood and urine tests, performed at his mother, promptly led us to the diagnosis of Toni Debré Fanconi syndrom family autosomal dominant [2,5]. In this context, the renal biopsy showed only discrete, non specific focal tubule interstitial lesions. But in this patient, a number of clinical and paraclinical elements, in particular chronic hepatosplenomegaly and thrombocytopenia, remained unexplained. They did discuss the diagnosis of lupus, but especially they led to the medullary biopsy and the demonstration of large foam cells leading to overload disease, Gaucher or Niemann-Pick disease, given the symptomatology in adult age of this patient [6]. Enzymatic studies [7] then genetic confirmed the diagnosis of NP syndrome linked to SMPD1 gene. The mutation observed in our patient is the homozygous delta R610 mutation, which is particularly common in Maghrebian patients [8,9] . The clinical picture associated with this mutation is that observed in type B [10], the most frequent and the least severe of the disease, and as in the cases reported in the literature, the diagnosis was late, at the age of 28 [8,11]. Cutaneous involvement has already been reported in the literature, characterized, as in our patient, by papular lesions of the face, back and thorax [12]. However, the presence of palpebral eodema has not been reported in the literature, which made us discuss a renal origin, this hypothesis quickly eliminated on the absence of important proteinuria, The radiological assessment revealed opacities suggestive of diffuse interstitial pneumonitis [5,11] and ultrasonography revealed heterogeneous liver parenchyma and presence of a hypoechoic nodule in the right adrenal lodge Interestingly, no cellular overload, no glomerulopathy was observed in the renal parenchyma, even during the directed reading of slides, after genetic confirmation of NP diagnosis. In the literature, two reports of renal impairment in patients with NiemannPick syndrome have been reported [13,14]. But in both cases, it was a fortuitous association, observed in patients with NP type C. On the other hand, an observation of renal damage directly related to the disease was reported in a patient with NP type A/B [15]. Impairment of renal function was detected at 14 years of age, more than 10 years after splenectomy. Renal biopsy revealed, apart from lesions of glomerular sclerosis and tubular atrophy, a massive lipid overload of podocytes, more irregular tubular cells associated with interstitial foci of foam cells. In electron microscopy, multiple, roughly lamellar, membraneembedded osmiophilic inclusions were present in the cytoplasm of podocytes, and more occasionally in vascular tubular and endothelial cells and sometimes in the nerves. These “myelinic” inclusions seems those observed in Fabry disease, but their distribution, respecting endothelial and mesangial glomerular cells, is different [16,17]. Unlike NP disease, renal impairment is commonly seen in other lipid storagerelated diseases, such as inherited lecithin-cholesterol-acyltransferase deficiency [18] or in other sphingolipidoses, such as Fabry’s disease or metachromatic leukodystrophy. In this patient aged 28 years, carrier of 2 pathologies, the long-term prognosis is reserved. The renal prognosis depends on the syndrome of Toni Debré Fanconi and therefore the risk of repeated episodes of dehydration leading, as in his parents, to calcium precipitation. A daily fluid intake, sufficient even in the young period, must prevent these accidents. On the other hand, NP disease exposes him to multiple complications, cardiac, hemorrhagic, hepatic, and pulmonary [8]. Apart from the symptomatic treatment, a substitution therapy will be indicating; a therapeutic approach that we think is necessary or indispensable in our country.
We report the exceptional association in one patient of two rare genetic pathologies, one autosomal dominant, the Toni Debré Fanconi syndrome , and the other autosomal recessive, Niemann-Pick disease. The diagnostic wandering was 24 years and to date; the patient receives only symptomatic treatment. This observation allows us to emphasize the importance of the problem posed by hereditary diseases in our country where there is a high rate of consanguinity [19], to insist on the need to develop the family and genetic studies essential to consider enzyme replacement therapies, and to promote genetic counselling to reduce the incidence of these severe hereditary diseases.
Dr. Ghalia Khellaf and the other authors declared no conflict of interest.
Download Provisional PDF Here
Article Type: CASE REPORT
Citation: Khellaf G, Saidani M, Missoum S, Rayane T, Kaci L, et al. (2019) A Syndrome Hides Another, About an Algerian Family. Int J Nephrol Kidney Fail 5(1): dx.doi.org/10.16966/2380-5498.170
Copyright: © 2019 Khellaf G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access