Introduction
Introduction The worldwide obesity epidemic has emerged as a major cause of cardiovascular disease, diabetes type II, and organ dysfunction, including kidney disorders [1]. Epidemiological studies indicate that obesity, particularly visceral obesity, puts individuals at risk of chronic kidney disease (CKD) [2-5].
In previous studies, we showed that low-density lipoprotein (LDL)-cholesterol overloading causes in renal tubular epithelial cells intracellular accumulation of cholesterol and phospholipids, particularly in lysosomal and autophagosomal organelles, with formation of large multilamellar bodies (MLBs) [6,7], which are a typical feature of lysosomal storage diseases (LSD) and phospholipidosis [8,9]. In addition to provoking destabilization and dysfunction of the lysosomal compartment, LDL loading also triggers mitochondrial damage, autophagy, and finally tubular damage and impairment of tubular absorption properties [7].
Membrane rafts are small, highly dynamic sterol-and sphingolipidenriched domains that provide platforms for protein-protein interactions and initiation of signaling cascades [10]. Therefore, they are important in compartmentalizing cellular processes and facilitating intercellular communication, as rafts concentrate many specific cell surface receptors, such as epidermal growth factor receptors (EGFR) [11,12].
Most cholesterol is transported in human blood in the form of LDL, which is organized in a core-shell structure. The lipid core contains primarily triacylglycerols, cholesterylesters (CE), and some unesterified free-cholesterol (FC), whereas the outer shell is constituted by phospholipids (mainly phosphatidylcholine and sphingomyelin), most of the free cholesterol and one single copy of apolipoprotein B-100 (ApoB-100), which is the principal ligand for the LDL receptor (LDLR) [13,14]. Once bound to the LDL receptor, LDL is rapidly internalized in clathrin-coated vescicles and delivered to early endosomes. Consequently, LDLR is recycled to the plasma membrane and LDL is trafficked via late endosomes to lysosomes, where acid hydrolases convert LDL-CE into free-cholesterol. As a result, freecholesterol becomes available for new membrane synthesis and is shuttled to other cell compartments such as endoplasmic reticulum (ER) and plasma membrane [15].
In presence of reactive oxygen species, native LDL (nLDL) is oxidized resulting in shedding of the polar outer layer, conformational changes of ApoB protein, and formation of new complex products, such as ceramide, oxysterols, oxidized phospholipids, and lysophospholipids. Due to ApoB unfolding and generation of novel immunogenic epitopes during LDL oxidation, oxidized LDL (oxLDL) is recognized mainly not by LDLR, but rather by innate immunity pattern recognition receptors (e.g. Toll-like receptor-TLR-4 and -2) and scavenger receptors (e.g. CD36, SR-BI, LOX-1) [13,16,17].
Elevated levels of oxLDL correlate with plasma hyper cholesterolemia [18-20] and are associated with an increased risk of coronary artery disease (CAD) and atherosclerotic plaque formation [21-23].
In this study, we focused on the impact of native and oxidized LDL on plasma membrane microdomains (membrane rafts) and on the downstream effect of altered membrane lipid raft composition.
Here, we show that in renal tubular epithelial cells (TEC) uptake of LDL particles, particularly when oxidized, alters plasma membrane lipid composition, affects cell proliferation, and epidermal growth factor (EGF)-induced signaling and EGF receptor availability at the plasma membrane. These data indicate that lipid loading alters cell membrane biophysical properties and thereby might despair the signaling function of membrane lipid/protein platforms.
Results
Oxidized LDL is a major inducer of cholesterol-and ceramide-rich plasma membrane microdomains
In previous studies, we showed that feeding mice a high-cholesterol Western-type diet caused ectopic lipid deposition within renal tubular epithelial cells [6,24]. Lipidomic analysis of renal tissues from mice subjected to western-type diet showed a significant increase in freecholesterol, fatty acids and several phospholipids as compared to kidneys of control diet-fed mice [6].
After uptake, via receptor-mediated endocytosis, LDL particles are trafficked to lysosomes, where acid hydrolases start the degradation of LDL-CE, -triglycerides, and -phospholipids. The free-cholesterol resulting from LDL lysosomal digestion is trafficked to other cell compartments including plasma membrane [15]. Therefore, in the first set of experiments we investigated the changes in cholesterol-rich microdomains (lipid rafts) in plasma membrane of cultured TEC using fluorescently tagged cholera toxin B subunit (CTB),which selectively binds to ganglioside GM1, a marker of lipid rafts [25-27].
Measurement by flow cytometry (FC) of cholesterol-lipid rafts revealed a time-dependent increment of plasmatic membrane cholesterol (day 3 and day 5) particularly after oxLDL uptake (Figure 1A).
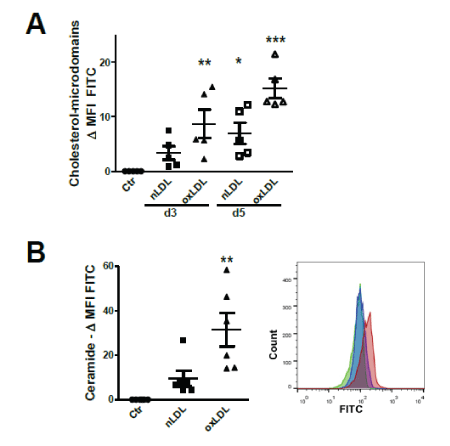
Figure 1: Plasma membrane alterations after nLDL/oxLDL loading.
(A) Flow cytometry (FC) data analysis of HK2 cells stained for cholesterol-rich plasma membrane microdomain with Cholera-toxin B (CTB)-FITC (FC). Mean fluorescence intensity (MFI) of LPDS-treated controls (Ctr) subtracted from the MFI of nLDL/oxLDL- treated cells.
(B) FC analysis of HK2 cells stained for cell surface ceramide domains with FITC-labelled secondary antibody. Differences in MFI compared to Ctr LPDS group. Histogram peaks of LPDS (green), nLDL (blue) and oxLDL (red) treated cells.
Assays at day 3 (A, B) and day 5 (A). Data presented as mean ± SEM; *P<0.05, **P<0.01, ***P<0.001. Each dot represents the average value of one independent experiment using LPDS/LDL isolated from one plasma donor.
Among the lipid species that ectopically accumulate in tissues of obese people, ceramide has gained much attention, being patho physiologically relevant for the impairment of insulin signaling and intracellular handling of glucose [28-30]. Ceramide, a second major lipid component of membrane rafts [10,31], accumulates either as a consequence of de novo synthesis from the condensation of palmitate and serine or from sphingomyelin (SPM) break down by action of acidic or neutral sphingomyelinase (aSMase /nSMase) [32].
As both nLDL and oxLDL contain quite high amounts of SPM and ceramides [13], we examined the presence of ceramide-rich plasma membrane microdomains following nLDL and oxLDL treatment. FC staining of cell surface ceramide revealed that oxLDL significantly increase plasma membrane ceramide content after 3 days of stimulation (Figure 1B). Thus, LDL, in its native and oxidized forms, alters plasma membrane microdomains.
LDL loading impairs TEC proliferation rate and signaling activation
Lipid rafts/microdomains are believed to function as platforms for ligand-receptor interactions by providing microenvironments enriched in receptors, lipids and protein effectors for signaling activation [33].
Cell cycle analysis showed that prolonged exposure to nLDL or oxLDL (5 days) inhibits TEC proliferation (Figure 2A). In contrast to control and nLDL-treatment conditions, 5-day treatment with oxLDL abolished the increase in proliferation rate induced by stimulation with EGF, (Figure 2B). Accordingly, EGF-induced signaling was also impaired after 5-day LDL exposure, as shown by the diminished phosphorylation rate of EGF receptor and of the downstream signaling molecules phosphoinositide 3-kinase (PI3K) (Figure 2C). To show causality, we used methyl-β-cyclodextrin (MβC) to reduce membrane cholesterol content. Upon nLDL loading for 5 days, MβC significantly increased the number of proliferating cells (Figure 2D). In addition, MβC-mediated membrane cholesterol depletion during nLDL treatment enhanced the availability of EGFR at the plasma membrane, assessed by cell surface staining of EGFR for FC (Figure 2E). These data indicate a negative effect of excessive membrane cholesterol-rich domains on cell proliferation and, possibly, on receptor-initiated signaling.

Figure 2: LDL exposure impairs cell proliferation and signaling abilities of tubular cells.
(A) Percentage of HK2 proliferating cells relative to control equal to 1; cell cycle analysis by flow cytometry.
(B) Cell proliferative response to EGF after nLDL/oxLDL loading; % proliferating cells relative to control (=1).
(C) Western blot showing the phosphorylation rate of EGFR and PI3K after 5-day exposure to nLDL/oxLDL and stimulation with or without EGF in the last 24 hours; β-actin used as loading control. Intensity normalized for control.
(D) Percentage of proliferating cells measured after 5 days of treatment with nLDL with or without MβC-dependent membrane cholesterol depletion. Mean fluorescence intensity (MFI) of controls subtracted from the MFI of nLDL/oxLDL-treated cells.
(E) Staining for cell surface EGF receptor on 5 days nLDL-treated HK2 cells in presence or not of MβC. Data shown as differences in MFI.
(A-E) Assays at day 5. Mean ± SEM; *P<0.05, **P<0.01, ***P<0.001. Dots are the average values from experiments with different plasma donors for LPDS/LDL isolation.
Uptake of oxLDL triggers apoptosis
Ceramide-enriched microdomains function as signaling platform involved in processes such as apoptosis [34]. Using Annexin V staining, we show that 3-day exposure to oxLDL triggered apoptosis of TEC (Figure 3A).
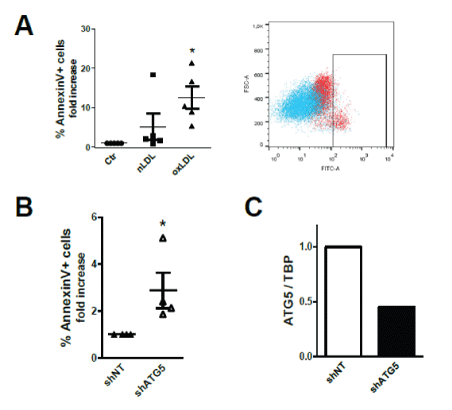
Figure 3: oxLDL triggers apoptosis.
(A) Fold-increase in % Annexin V-FITC+ apoptotic cells relative to control equal to 1. Scatter-plot showing Ctr (blue) and oxLDL-treated (red) HK2 cells.
(B) Apoptosis in oxLDL-treated cells stably expressing shRNA targeting ATG5. Fold-change in % Annexin V-FITC+ in ATG5 knock-downed cells compared to cells expressing non- targeting (NT) shRNA.
(A, B) All assays performed after 3 days of loading. Data shown as mean ± SEM; *P<0.05, **P<0.01, ***P<0.001. Each dot is the average from one independent experiment with distinct plasma donors.
(C) QPCR analysis of ATG5 gene expression in HK2 cells stably expressing lentiviral constructs encoding shRNA targeting ATG5 expression or nontargeting (NT) shRNA.
As in our earlier study, we disclosed that LDL loading also triggers autophagy in TEC [7], we used cells stably expressing short hairpin (sh)RNA targeting ATG5 to suppress the intracellular autophagic flux. ATG5 knock-down rendered TEC more susceptible to oxLDLinduced apoptosis (Figure 3B), suggesting that autophagy represents a protective mechanism during metabolic overload possibly adopted by cells to eliminate the disrupted lysosomes and to stop the leakage of their content in the cytoplasm [35,36], thereby diminishing apoptosis induction. Figure 3C shows the knock-down of ATG5 expression as compared to cells expressing non-targeting (NT) shRNA.
Discussion
We and others have proven a causal link between over nutrition and the appearance in kidneys of damage, inflammation and fibrosis, which altogether feature CKD [6,24,37-42]. In this study, we show a novel aspect of renal lipotoxicity at the cellular level of tubular epithelium which plays a major role in renal physiology.
We report an increase in plasma membrane cholesterol and ceramide content in TEC after nLDL/oxLDL exposure that appears to alter proliferation and signaling in response to EGF. Prolonged nLDL/ oxLDL loading drops the proliferation rate of tubular cells and impairs their response to epidermal growth factors in terms of proliferation and signaling. Strikingly, oxLDL abolishes the ability of EGF to augment the proliferation rate of TEC after 5 days of treatment. In addition, LDL uptake negatively impacts the EGF-mediated intracellular signaling, as assessed by the lower rate of phosphorylation of EGF receptor and of PI3K.
After uptake and transport to the lysosomes, LDL-cholesteryl esters are degraded into free cholesterol, which is eventually shuttled to other sites of the cell, such as the plasma membrane. As more and more cholesterol is trafficked to the plasma membrane following LDL lysosomal break-down, the increased plasma membrane cholesterol content can disrupt lipid-raft composition (receptors/lipid/signaling proteins) and presumably inhibit the trafficking/turnover of EGFR, which is localized in cholesterol-enriched membrane domains [12]. As a result, proliferation and signaling in response to EGF are altered after nLDL/oxLDL loading. In agreement, in 3T3 cells cholesterol levels were shown to modulate EGFR-mediated signaling; Pike et al. reported that membrane cholesterol depletion with MβC greatly enhances surface EGF binding and EGFR autophosphorylation [43]. Accordingly, we found that membrane cholesterol depletion by MβC enhances cell replication and the availability of EGFR at the plasma membrane.
Therefore, the consequences of lipid loading are not limited to alteration of the biophysical properties of plasma membrane but are likely extended to cellular function by displacing clustered proteins from lipid rafts.
Saturated fatty acids and ceramides are constituents of LDL particles and they are released after lysosomal LDL breakdown. Ceramide is a central intermediate in sphingolipid metabolism that is increased during diabetes and obesity [30,44]. The excessive uptake of saturated fatty acids upon nutrient overloading may in fact lead to de novo ceramide generation from palmitate by action of serine palmitoyltransferase [30,44]. Ceramide was shown to contribute to obesity-related disorders by promoting inflammation as well as insulin resistance mainly through inhibition of the protein kinase B PKB/Akt, a central mediator of insulin pathway [45]. Indeed, plasma and tissue levels of several ceramide species correlate with insulin resistance [30].
The formation of ceramide enriched raft domains after oxLDL uptake was previously described in macrophages [27]. Cell membrane ceramide-rich platforms can generate from sphingomyelin/ceramide conversion by active sphingomyelinases (SPMase) and oxLDL is known to prompt SPMase activity [27,46,47]. The excessive saturated fatty acid uptake upon nutrient overloading may also lead to de novo ceramide generation from palmitate by action of serine palmitoyltransferase [30,44]. Fatty acids and ceramide are contained in the LDL particles and released intracellularly after LDL lysosomal breakdown. In response to stress-stimuli (i.e. inflammation, TNF-α, and IL-1), ceramide mediates many diverse cellular pathways including apoptosis, cell senescence, cell proliferation arrest, intracellular trafficking, insulin resistance and inflammation [32,48-51].
In tubular cells, mostly oxidized LDL could provoke apoptosis. Considering that only oxLDL could enhance ceramide plasma membrane levels, and in view of the role of ceramide platforms in apoptosis, it is reasonable to hypothesize a causal relationship. In endothelial cells, oxLDL-derived neutral lipids, such as oxysterols, were found responsible for the oxLDL apoptosis-inducing activity through generation of ceramide by membrane sphingomyelinase activation [52]. In our previous study, we disclosed that exposure of tubular epithelial cells to LDL induces lysosomal phospholipid accumulation and hence triggers lysosomal disruption due to lysosomal overloading of undigested lipid cargo [7]. The release of endolysosomal contents into the cytoplasm is a trigger of apoptosis and, at early stage, of autophagy [8,35,53].
In conclusion, alteration of lipid content of plasma membrane may disrupt signaling by displacing receptor or signaling effect or proteins normally localized within or linked to cholesterol raft membrane domains and thus compromise cell communication and response to growth factors fundamental in repair processes. This might be particularly relevant for allograft survival after ischemia/reperfusion injury due to transplantation. Indeed, obesity and metabolic syndrome are risk factors for allograft loss and poor kidney function in kidney transplant recipients [54-56].
Methods
Isolation of LDL and generation of oxidized LDL
A detailed description of LDL isolation and oxidation protocols is provided in Supplementary Information in [7]. Briefly, LDL particles were isolated from 300-400 ml plasma of healthy donors by sequential KBr-density gradient ultracentrifugation (UC) steps (rotor 70Ti, Beckman). Potassium bromide was added according to this calculation KBr gr=Volume plasma ml × 0.019 and after the first UC (58000 rpm 22h 4°C) the top fractions containing very-low density and intermediate-density lipoproteins (VLDL, IDL) were discarded. Subsequently, KBr was added as follow KBr gr=Volume ml × 0.066; after the second UC (58000 rpm 22h 4°C) the upper fraction of LDL was harvested. The remaining plasma was centrifuged two more times (48000 rpm 48h 4°C) for isolation of lipoprotein-deficient serum (LPDS in bottom phase). Both LDL and LPDS were dialyzed (using Servapor Dialysis Tubing) in 3l PBS/EDTA for 3 times (2h each) and afterwards in 3l PBS (3 × 2h each).
To oxidize LDL, CuSO4 was used: LDL fractions were dialyzed at a concentration of 1mg protein/ml against 3l PBS containing 5 µM CuSO4 for 36h at 4°C. Subsequently oxLDL was dialyzed against PBS/ EDTA and PBS [7, 57].
Healthy plasma donors were selected on basis of healthy blood glucose (60-100 mg/dl) and blood lipid levels (cholesterol 60-200 mg/dl, high-density lipoprotein HDL 35-55 mg/dl, LDL 30-150 mg/ dl, lipoprotein A <30 mg/dl, triglycerides 20-200 mg/dl), body mass index (BMI<25), ApoE3/E3 genotype and age (<45 years); written informed consent was obtained by each donor as previously described [7]. The Ethical Committee of the University of Regensburg, Germany approved all procedures.
Cell culture and assay procedure
HK2 renal tubular epithelial cells were cultured as previously described [7]; in vitro assays lasted for 3 or 5 days: cells were culture in Dulbecco’s Modified Eagle Medium DMEM (Gibco) containing lipoprotein-deficient serum (LPDS, control) or 5 µg/ml nLDL or oxLDL [7]. Medium was replaced every second day. EGF (20 ng/ml, R&D Systems) stimulation was done in the last 24h of 5 days long experiments. Methyl-β-cyclodextrin (5 mM, Sigma-Aldrich) was utilized to deplete membrane cholesterol for 30 min at day 2 and 4 in 5 days lasting experiments.
Gene silencing
Stable knockdown cells were generated by transduction with lentiviral particles harbouring shRNA in presence of 8 µg/ml polybrene (Sigma Aldrich) for 24h; transduced cells were selected with 10 µg/ml puromycin (Sigma Aldrich) 48h after transduction. Lentiviral particles were produced by transfecting HEK293T cells, using GENIUS DNA Transfection Reagent (Westburg), with pMD2.G/VSVG (Addgene 12259), pPAX2 (Addgene 12260) and the lentiviral pLKO.1 shRNA vector (Addgene 10878) at a 6:15:20 µg/ DNA ratio. To suppress autophagy, the pLKO.1 vector (Addgene 10878) containing an shRNA insert targeting human (h) ATG5 (TRCN0000150940) was used.
Staining for flow cytometry analysis
Flow cytometry staining of plasma membrane cholesterol, ceramide, EGFR, phosphatidylserine and of nuclear DNA was visualized on a FACSCanto II (BD Biosciences); data analysis was performed using the FlowJo software (TreeStar). Cell surface cholesterol-rich domains were stained with FITC-tagged cholera toxin B subunit (2.5µg/ml, SigmaAldrich), which binds to GM1 ganglioside present in lipid rafts [25- 27]. Plasma membrane ceramide was stained with mouse monoclonal antibody anti-ceramide IgM (1:20, clone MID 15B4, Alexis) followed by goat anti-mouse IgM Alexa Fluor 488-conjugated (Invitrogen Molecular Probes). Cell surface EGFR expression was detected by anti-EGFR (Cell Signalling Technologies) staining followed by Alexa Fluor 488-conjugated anti-rabbit IgG (Invitrogen Molecular Probes). For measurement of apoptotic cells, Annexin V staining of surface phosphatidylserine in Annexin V binding buffer (BD Pharmingen™ Apoptosis Detection Kit) was performed. Cell cycle analysis was assessed after over-night ethanol fixation by staining DNA in a PBS solution containing 10 µg/ml propidium iodide (Invitrogen) and 250 µg/ml RNAse A (Macherey-Nagel).
Gene expression analysis
Total RNA was extracted from cells with Trizol reagent (Invitrogen). RNA was converted to cDNA by using oligo-dT primers. SYBR GreenSensiMix (Bioline) was used for quantitative real-time PCR (QPCR) on a LightCycler® 480 System (Roche). Linear regression analysis was used for analysis of SYBR green dye intensity and gene expression was normalized towards the housekeeping genes TATA-box binding protein (TBP). Oligonucleotide primer sequences (5’→3’) for each gene are:
TBP forward CAGGAGCCAAGAGTGAAGAAC reverse GGAAATAATTCTGGCTCATAGCTACT, ATG5 forward GTTTGTCCTTCTGCTATTGATCC reverse TCAGATGTTCACTCAGCCACT.
Statistics
Statistical analysis was performed using One-Way ANOVA and Dunnett’s tests or Mann-Whitney U test used for two group comparison; P<0.05 was considered to be significant. Data are presented as mean and standard error of the mean (SEM). In dot-plot graphs, each dot represents the average of one independent experiment using LPDS/LDL isolated from one plasma donor.