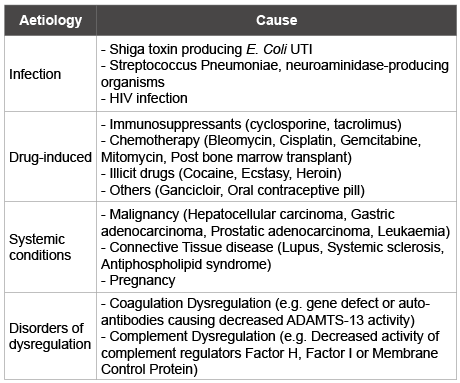
Table 1: Causes of atypical HUS
Hannah Wilkinson* John Dixon Fiona Harris
South West Thames Renal and Transplantation Unit, St. Helier University Hospital, Carshalton, Surrey, UK*Corresponding author: Hannah Wilkinson, Nephrology Department, St. Helier Hospital, Wrythe Lane, Carshalton, Surrey, United Kingdom, Tel: 07967791995; E-mail: hannah.wilkinson@esth.nhs.uk
Background: Atypical HUS (aHUS) occurs due to endothelial damage following abnormal complement regulation, with some cases being deficient in complement regulating factors H, I and Membrane Cofactor P.
Case presentation: We present a case of a 28 year old female presenting with flu like illness, mild renal impairment, haematoproteiunurea and anaemia. Renal biopsy showed widespread endothelial swelling and multiple red cell and platelet fibrin thrombi occluding the vessel lumens and a diagnosis of aHUS was made. She was found to have persistently low C3 with a normal C4 and functional complement assays revealed Factor I deficiency. She was successfully treated with plasma exchange. In contrast to other reported cases of Factor I deficiency-associated aHUS our patient has achieved good renal recovery and remains dialysis free at 4 years. Only a handful of cases of Factor I deficiency associated with a HUS have been reported; the aetiology of this case was thus a diagnostic challenge.
Conclusion: The authors recommend that complement regulator disorders are considered in aHUS and that persistently abnormal laboratory results are investigated further if the aetiology is unclear.
Complement factor deficiency; HUS
aHUS: Atypical HUS; ANA: Antinuclear Antibody; ANCA: Anti-neutrophil Cytoplasmic Antibodies; D+ HUS: HUS with Diarrhoea; D-HUS: HUS without Diarrhoea; LDH: Lactate Dehydrogenase; MCP: Membrane Cofactor Protein; RBC: Red Blood cells; RCA: Regulators of Complement Activation.
Haemolytic Uraemic Syndrome (HUS) is a rare disorder characterised by microangiopathic haemolytic anaemia, thrombocytopaenia, acute renal impairment, and hypertension. HUS predominantly affects children and is associated with diarrhoea in the majority of cases (D+ HUS). HUS that has a non-diarrhoeal aetiology is termed atypical (D-) HUS. Atypical HUS accounts for 5-10% of all cases [1]. D+HUS is usually preceded by Shigella or E. Coli 0157:H7 infection causing Shiga-toxin production. This then causes diarrhoea and endothelial cell wall damage, resulting in HUS [2]. Atypical HUS has been associated with several conditions that also result in endothelial cell injury (Table 1). It has recently been demonstrated that atypical HUS is caused by altered regulation of complement [3] with approximately half of all cases being due to deficiencies in the complement regulating proteins Factor H, Factor I and Membrane Cofactor Protein (MCP)[4].
Table 1: Causes of atypical HUS
Atypical HUS often provides a diagnostic challenge and has a poorer prognosis with death rates of 25% in the acute phase and 50% needing ongoing renal replacement therapy [1]. We present here a case of atypical HUS due to complement factor I deficiency that was successfully treated with plasma exchange.
A 28 year old woman presented with a four week history of a flulike illness and a headache. There were no other specific symptoms, in particular, no diarrhoea or neurological symptoms. She had been diagnosed as having coeliac disease at three years of age, and had been well controlled on a gluten-free diet. During the previous three years she had had three separate episodes of acute facial swelling, lasting a few days, with no apparent trigger. She also gave a history of recurrent sore throats with one episode associated with a positive throat swab for beta haemolytic Streptococcus. Previous blood pressure recordings had always been normal and she had never been pregnant She was not taking regular medications and gave no history of ‘over the counter’ drug use or herbal remedies. Her family history was significant in that her sister died aged three years of age from dialysis dependent renal failure, believed to be due to Haemolytic Uraemic Syndrome. Her other sister and parents had no significant health problems. Clinical examination revealed a blood pressure of 150/90mmHg but no features to suggest malignant hypertension. In addition, there were no features to suggest scleroderma. Examination was otherwise unremarkable. Dipstick urinalysis showed haematuria (2+) and proteinuria (3+), which was later quantified at 3 g/24 hours. Initial investigations revealed the mild renal impairment (urea 8.7 mmol/L, creatinine 110 µmol/L), mild anaemia (Hb. 10.6 g/dL), and a mildly elevated LDH (336 U/L). Other haematological and biochemical investigations were within normal limits: WCC 5.4x109 /L, Platelets 298, Bilirubin 17, AST 25 U/L. Some RBC fragments and occasional spherocytes were seen on her blood film. Immunological serology was negative for ANA, ANCA, and Autoantibodies. Rheumatoid Factor, Cryoglobulins and antiphospholipid antibodies were undetectable. Of relevance, her C3 was low at 0.73 (NR 0.75-1.65), and C4 normal at 0.28 (NR 0.14-0.54). Given our initial findings, we went on to perform a renal biopsy which showed widespread endothelial swelling and retraction. Multiple red cell and platelet fibrin thrombi occluding the vessel lumen.
At the time our differential diagnoses were; Malignant hypertension but the Blood Pressure was not sufficiently abnormal, Scleroderma but there were no abnormal serology or suggestive clinical features, Pre-eclampsia but she was not pregnant, Anti-cardiolipin related disease Antibodies were not present and early HUS.
Given the clinical and laboratory findings, a diagnosis of HUS was made. She was treated with five cycles of plasma exchange using Fresh Frozen Plasma. It was noted at subsequent outpatient clinic appointments that C3 levels were persistently low, with normal C4 levels. In view of this, functional complement assays were performed. Normal results during CH100 (972 units; range: 488-1150) and AP100 (145 units) testing were obtained, thus implying that all components of the classical and alternative complement pathways, respectively, were intact. Factor H level was 0.5 mg/L (range: 0.2-0.6), and Factor I level was low at 0.32 mg/L (range: 0.4-0.6). This complement profile is consistent with uncontrolled alternative pathway activation and is consistent with heterozygous Factor I deficiency.
Recent work has found that atypical HUS occurs as a result of endothelial damage due to abnormal complement regulation [3]. Approximately half of all cases are due to deficiencies in either of the complement regulating proteins Factor H, Factor I or Membrane Cofactor Protein [4]. Factors H and I are synthesised by the liver, whereas MCP is a transmembrane cell regulator appearing on many cells.
Factor H inhibits the formation of alternative C3-convertase and accelerates its decay. C3-convertase acts to enzymatically convert C3b, the penultimate product in the alternative complement pathway, to C3, the final product. Factor H and Membrane Co-factor Protein are co-factors for the C3b–cleaving protease Factor I. By cleaving C3b the pathway can no longer continue [3]. Inhibition or deficiency of any of the complement regulating proteins leads to over-stimulation of the complement cascade. It has been suggested that an external trigger induces endothelial cell injury and activation of the complement cascade [3,5]. These exposed cells now require protection from the effects of complement. If deficiencies of the complement regulating proteins occur then unregulated complement action ensues. A pro-coagulant state is induced, thus inducing the process of thrombus formation in the microvasculature.
Several studies have reported mutations in genes controlling the alternative complement pathway control proteins Factor H, Factor I, and MCP [5-7].Genetic analysis has localised genes for Factor H and MCP to an area on the long arm of Chromosome 1 at locus 1q32 called the Regulators of Complement Activation (RCA) gene cluster, and genes controlling Factor I to Chromosome4 at locus 4q25 [4,8]. More than fifty different abnormalities have now been reported in the Factor H gene. Many of these changes have been overrepresented in patients with Factor H when compared with normal controls. In a recent study comparing Factor H-gene knockout mice with controls each of the transgenic mice developed spontaneous HUS between six and twelve months later and C3 deposition in glomeruli and renal vasculature was seen after three weeks [9]. This suggests that an active complement system and defective protection of the renal endothelium is necessary for the development of atypical HUS. A single case report has revealed atypical HUS associated with low Factor H levels due to anti-Factor H antibodies [10].
Only a handful of cases of Factor I deficiency associated with atypical HUS have previously been reported in the literature [6,8,11]. Several changes that are seen in patients with HUS due to Factor I deficiency have not been detected in controls. This case, along with three of the previously described cases, probably has a hereditary component. Two additional studies report the case of eight patients with mutations in the Factor I protein that developed atypical HUS. None of these patients have a reduction in the level of Factor I or in its activity [7,9].
In contrast to our case, previously described atypical HUS associated with Factor I deficiency has had a poor prognosis. Four cases have subsequently developed end-stage-renal-failure, and HUS recurred in the three that received renal transplants. HUS recurred in the fifth case within six months. No information is available for the remaining documented case. Our patient has not had a recurrence after four years following initial therapy with plasma exchange.
There are no randomised controlled trials about the efficacy of plasma exchange in atypical HUS, however, mortality rates have dropped from 50% to 25% following the introduction of plasma exchange [4]. It has been postulated that this is due to the removal of toxic substances. Renal transplantation for atypical HUS associated with MCP deficiency has a better prognosis than Factor H or Factor I deficiency; recurrence rates being 10%, 80% and 100% respectively [2]. This is likely to be due to the fact that Factors H and I are synthesised in the liver and are thus not replaced by renal transplantation, whereas MCP is expressed as a transmembrane regulator and will thus be present in donated kidneys.
This was a challenging case due to its several differentials and the rarity of its aetiology. The main differential was that of malignant hypertension, however, there were no clinical features to suggest this, and her blood pressure was not high enough to explain the biopsy findings. Scleroderma was another possibility, but again there were no such suggestive clinical features. The other differentials of the renal biopsy were pre-eclampsia, antiphospholipid syndrome, and early HUS. She was not pregnant, anti cardiolipin antibodies were not detected, and laboratory features of HUS were not typical. The main clue to the aetiology was the persistently low C3 level with a normal C4 level. This, combined with normal intrinsic complement component assays, suggested an abnormality in one of the complement regulating proteins.
While rare, complement regulator disorders represent an important diagnosis to consider when faced with D-HUS. The authors would recommend that complement regulator disorders are considered in HUS without diarrhoea. In addition, we would recommend that persistently abnormal laboratory results are investigated further if the aetiology is unclear.
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images.
The Authors declare that there is no conflict of interest.
HW and JD designed, researched and wrote the article.
HW was the primary author.
FH assisted with research and edited the article prior to submission.
Each author approves the submitted version.
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief on request. The authors gratefully acknowledge the investigations performed by Professor T. Goodship, Institute of Human Genetics, and University of Newcastle upon Tyne, United Kingdom, that diagnosed the Factor I deficiency in this patient. The authors acknowledge Dr. S. Samson, Department of Histopathology, Epsom and St. Helier University Hospital, Carshalton, Surrey, UK for providing the renal biopsy photographs.
Download Provisional PDF Here
Article Type: Case Report
Citation: Wilkinson H, Dixon J, Harris F (2016) Case Report: Atypical Haemolytic Uraemic Syndrome Due to Abnormal Alternative Complement Pathway Regulation. Int J Nephrol Kidney Failure 2(3): doi http://dx.doi.org/10.16966/2380-5498.131
Copyright: © Wilkinson H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access