Abstract
Lithium is a useful treatment for bipolar disorder; it attenuates the recurrence of affective episodes and reduces the risk of suicide among
bipolar patients. Nevertheless, lithium has a plethora of side effects some of which are serious and may be irreversible. Among the most troubling
side effects of lithium is its nephrotoxicity. Herein we report a case of a bipolar patient who developed signs of lithium-induced nephrotoxicity
including overt polyuria after 18 years of lithium treatment. Due to concerns that his renal function will continue to deteriorate, two trials to switch
him to other psychotropic drugs were done following which he committed two aggressive suicidal attempts. As a result, a joint decision was
reached between the patient, his family, psychiatrist and nephrologist that lithium is a “life-saving” treatment for him and lithium was never stopped
again. To date, at 68 years old and after nearly 40 years of lithium treatment the patient is mentally stable and reasonably functional. His kidney
function continued to deteriorate slowly through the years and he is now a candidate for hemodialysis. This case report emphasizes the need
to balance between concerns regarding the damage to the kidney which is a result of long-term lithium treatment to the significant therapeutic
benefit of lithium as a mood stabilizer and anti-suicidal drug.
Keywords
Bipolar disorder; Creatinine; Lithium; Polyuria; Suicide
Introduction
Lithium is the gold standard treatment for bipolar disorder [1,2]. It
attenuates the recurrence of affective episodes [1,2] and reduces suicidal
attempts and suicidal death among bipolar patients [3,4]. However,
despite its established therapeutic efficacy, long-term lithium treatment is
complicated by two limitations: (i) lithium has a narrow therapeutic index
and increased risk of intoxication, and, (ii) lithium has several side effects,
some of which are severe and occasionally irreversible [5-7].
Lithium is a simple chemical that does not undergo hepatic
metabolism. Nevertheless, addressing its therapeutic and toxicological
profiles necessitates acknowledgement of the variability among patients in
their response to the drug. The therapeutic response and the toxicological
profile of lithium may greatly differ among patients even at similar plasma
levels, due to inter-individual variations in sensitivity to the drug and
differences in renal excretion [2,5,6,8].Patients can differ in their response
to lithium due to different age, gender, ethnic background, comorbidities,
interaction with other medications and genetic variations [2,5,6,8].
Plasma levels do not always predict the severity of lithium intoxication
despite being a principal factor that guides the clinical assessment of
patients. Some patients develop signs of lithium toxicity while having
plasma concentrations that are within the recommended therapeutic
range [9]. Thus, assessment of a patient’s situation must mainly rely on
the clinical presentation and severity of symptoms [10]. Several factors
may affect plasma lithium levels and the risk for toxicity. Impaired renal
function is one of the major factors that may lead to lithium intoxication
due to decreased elimination and accumulation of the drug [10-12].
Many drugs may influence lithium clearance by altering renal blood
flow, glomerular filtration rate (GFR) and sodium balance. For example,
Non-steroidal Anti-inflammatory Drugs (NSAIDs) are known to reduce
lithium clearance, resulting in elevated plasma concentrations of the
drug [10-12]. These drugs inhibit the Enzyme Cyclooxygenase (COX)
and thereby diminish prostaglandins (PGs) production. Inhibition of
PGs synthesis reduces renal blood flow and GFR, the result of which is
enhancement of lithium reabsorption [10-12]. The interaction between
lithium and NSAIDs occurs with classical NSAIDs as well as selective
COX-2 inhibitors. However, this interaction seems to be less frequent
with aspirin than other NSAIDs [11,12]. Angiotensin Converting Enzyme
(ACE) inhibitors may increase the risk for toxicity because they reduce
glomerular perfusion pressure (due to dilatation of efferent arterioles)
and decrease lithium glomerular filtration [10-12]. Furthermore, medical
conditions that are associated with volume depletion and diminished
renal blood flow such as diarrhea and sepsis may reduce GFR and increase
the risk of lithium toxicity.
One of the most worrisome side effects of lithium is impairment of
kidney function. Potential deleterious effects of lithium on renal function
include a decrease in urinary concentrating capacity and a reduction
in glomerular filtration rate, among other complications [5-7,13-20].
The most common renal side effect of lithium is Nephrogenic Diabetes
Insipidus (NDI) [5,7]. Important factors that contribute to the development
of lithium-induced NDI are: increased blood lithium levels, long duration
of treatment and high incidence of lithium intoxication episodes [5-
7,13-20]. Importantly, NDI may appear even after lithium cessation [21]
and withdrawal of lithium does not necessarily reverse the impairment
in urinary concentrating ability [7,16,17,22]. This article presents a case
of a patient who suffered several episodes of lithium-induced NDI and
gradually developed a chronic kidney disease. Thereafter, it discusses the
pathophysiological mechanism(s) underlying lithium-induced NDI.
Case Report
A male patient started to receive lithium at the age of 28 years after being
diagnosed as having bipolar disorder. Lithium treatment was continued
for nearly 18 years during which plasma lithium concentrations where
within the therapeutic range (0.6-1.2 mEq/L) and no documentation of
lithium intoxication episodes. At age 46 years the patient was referred
to a nephrologist (A.S.) due to complaints on dry mouth, polydipsia,
polyuria and deterioration in renal function. His clinical assessment at
the nephrology clinic revealed the following findings: blood pressure was
normal; plasma creatinine =1.4 mg/dl, urea =34 mg/dl, sodium =139
mEq/L; eGFR =57 ml/min; urine output =8 L/day, without proteinuria,
glucosuria, red blood cells or casts; kidney ultrasound was normal. After
an initial examination, a water deprivation test was performed revealing
the following results: before the test – plasma osmolality =283 mOsm/
kg, urine osmolality =164 mOsm/kg; 6 hours after water deprivation –
plasma osmolality =298 mOsm/kg, urine osmolality =165 mOsm/kg; after
administration of vasopressin – urine osmolality =174 mOsm/kg. After
the test, a diagnosis of lithium-induced NDI was made and lithium was
stopped and the patient was switched to other psychotropic drugs (such as
valproate, carbamazepine, antipsychotics). The cessation of lithium did not
alleviate the symptoms of NDI and after 2 months the patient attempted
suicide and was hospitalized for several weeks. During hospitalization
lithium was reinstated and the patient became affectively stable (euthymic)
and resumed his job. After 5 years (age~51 years), lithium was stopped
due to further deterioration in kidney function (plasma creatinine =1.7
mg/dl, eGFR =47 ml/min). The patient was treated with other mood
stabilizers for few months after which he committed a suicide attempt
once again and was hospitalized. A joint meeting was conducted between
the treating psychiatrist, nephrologist, patient and his family – and it was
decided to reinstate lithium despite the possibility of further deterioration
in NDI and renal function. It was also agreed upon that lithium is a “lifesaving”
treatment for the patient and that it will not be stopped again
even if it will lead to renal replacement therapy. Today, after nearly 40
years of treatment with lithium (excluding two pauses of few months)
and 21 years of a nephrologist follow-up, the patient is mentally stable
and reasonably functional. The dose of lithium is adjusted according to its
plasma concentration. His renal data is as follows: plasma creatinine =4.1
mg/dl, eGFR =16 ml/min; urine output =6-7 L/day, proteinuria =800 mg/
day; renal ultrasound – echogenic kidney with decreased corticomedullar
differentiation and cortical cysts; the patient is on the waiting list for
hemodialysis.
Mechanism of Lithium-induced NDI
The pathophysiological mechanisms underlying lithium-induced NDI
are not clearly understood. Understanding the physiological processes
involving renal sodium and water homeostasis is a key point. In the
kidney, on the apical membrane of proximal tubules and principal cells of
the collecting duct the major proteins that transport lithium into cells are
the sodium-hydrogen exchanger (NHE) and the epithelial sodium channel
(ENaC), respectively (Figure 1) [10,23-25]. It is established that lithium
can substitute for sodium and enter cells through sodium-transporting
systems [10,23-25] particularly during states of dehydration and volume
depletion. There are other transporting systems that may transfer lithium
into tubular cells such as the sodium-phosphate cotransporter(Figure 1)
[10,23-25], however, their relevance to the entry of lithium into kidney
cells is still unknown. On the basolateral side of proximal and distal tubules,
the most likely possibility for lithium to be extruded out of cells (to the blood)
is through the sodium-sodium exchanger (Figure 1) [10,23-25].
The transport of water into renal tubular cells is mediated through
the water channel protein aquaporin (AQP) (Figure 1) [26]. AQP1 is
abundantly expressed in the proximal tubule and descending loop of
Henle where it is localized to the apical and basolateral membranes [26]. In
the collecting duct, the transport of water into principal cells is mediated
mainly by AQP2 (Figure 1) [27]. The exit of water occurs through AQP3
and AQP4. Normally, water permeability of principal cells is regulated by
vasopressin. Vasopressin activates V2 receptors and increases intracellular
cyclic adenosine monophosphate (cAMP), leading to translocation
of AQP2 from intracellular vesicles to the apical membrane [27,28].
Prostaglandins also play a role in the regulation of sodium and water
reabsorption [29]. PGE2
is the main prostaglandin synthesized in the
kidney [29] and its effect on renal function seems to be site-specific [29-
31]. For example, in the collecting duct, PGE2
attenuates the antidiuretic
effect of vasopressin [31].
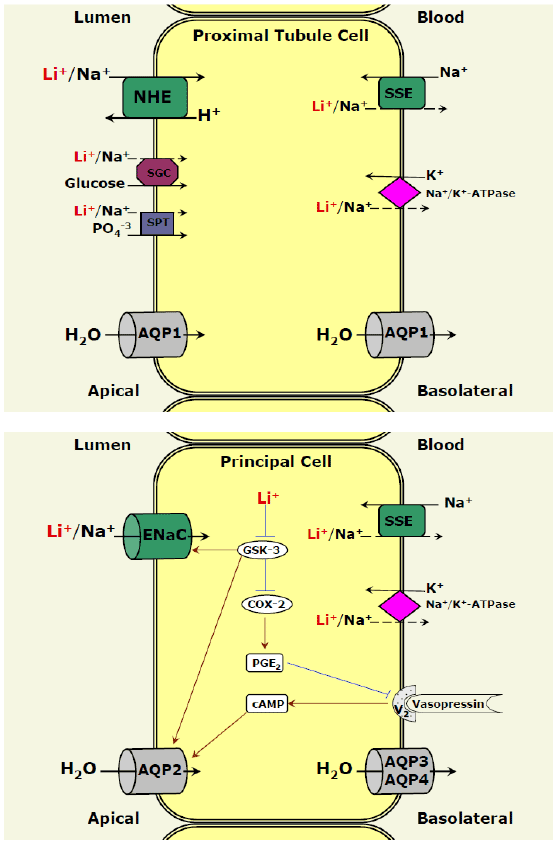
Figure 1: Lithium transport in kidney cells. In proximal tubule cells
(top panel) and principal cells of the collecting duct (lower panel) lithium
may substitute for sodium and be transported via sodium-transporting
systems. Lithium is transported into proximal tubule cells and principal
cells through NHE and ENaC, respectively. Other pathways for
entry of lithium are less likely and may include: the sodium-glucose
cotransporter, the sodium-phosphate transporter and the sodium-amino
acids cotransporter. These pathways may be particularly active during
states of dehydration and volume depletion. Extrusion of lithium to
the blood may occur through SSE or Na+/K+-ATPase. Lithium inhibits
GSK-3 leading to increased COX-2 expression and PGE2
synthesis.
This results in diminished vasopressin activity and decreased AQP2
levels on apical membrane of principal cells, which leads to increased
urination. Abbreviations: AQP, aquaporin; COX-2, cyclooxygenase 2;
ENaC, epithelial sodium channel; GSK-3, glycogen synthase kinase 3;
NHE, sodium-hydrogen exchanger; SSE, sodium-sodium exchanger;
PGE2
, Prostaglandin E2
; SGC, sodium-glucose cotransporter; SPT,
sodium-phosphate transporter.
Lithium decreases the antidiuretic effect of vasopressin after short as
well as long-term treatment duration [32,33]. The mechanisms by which
chronic lithium therapy may reduce the antidiuretic effect of vasopressin
are: First, lithium enhances PGE2
production, which decreases
vasopressin-induced cAMP synthesis [33]. Induction of COX-2 is a
crucial mechanism by which medullary interstitial cells adapt successfully
to the rapid shifts in ambient tonicity normally occurring in renal medulla
[34]. These adaptive mechanisms are partially regulated by the enzyme
glycogen synthase kinase 3β (GSK-3β), which is regarded as an up-stream
modulator of COX-2 expression [34]. Lithium inhibits GSK-3β [35,36],
which results in increased expression of COX-2 (i.e., GSK-3β negatively
regulates COX-2 expression) [34]. Thus, the regulation of renal sodium and
water homeostasis by the GSK-3β-COX-2 pathway may be summarized as
follows: (i) PGE2
increases urination by attenuating the antidiuretic action
of vasopressin; (ii) GSK-3β enhances the antidiuretic action of vasopressin
by decreasing COX-2 expression and reducing PGE2
synthesis; (iii)
lithium inhibits GSK-3β leading to increased COX-2 expression and
PGE2
synthesis the result of which is diminished vasopressin activity
and increased urination (Figure 1). These understandings were probably
the basis for using NSAIDs and COX-2 inhibitors as a treatment against
lithium-induced NDI [27,37,38]. Second, lithium reduces AQP2 gene
transcription through a PG-independent mechanism, leading to further
decrease in urinary concentrating ability [39]. Third, lithium induces
remodeling of collecting duct which characterized by a decrease in the
number of principal cells and an increase in the number of intercalated
cells [40]. Fourth, lithium was found to decrease the ratio between
principal and intercalated cells in mice collecting duct due to G2 (cell
cycle) arrest in principal cells [41]. The decrease in principal/intercalated
cell ratio was accompanied with features of NDI.
The management and treatment of a patient with lithium-induced
NDI should take into account several important factors including:
severity of NDI, risk of affective deterioration if lithium is stopped,
stage of tubulointerstitial damage (if the damage is irreversible the
patient will not necessarily benefit from the cessation of lithium), and,
availability and feasibility of treatment options (for example, if a severe
NDI develops due to acute lithium intoxication, hemodialysis should be
immediately considered to minimize the damage to the kidney). Several
pharmacological interventions have been suggested as a treatment for
lithium-induced NDI. The potassium-sparing diuretic amiloride is one of
the established options [15,42,43]. Other options are thiazide diuretics and
vasopressin; however, their efficacy and safety remain to be ascertained.
Classical NSAIDs and selective COX-2 inhibitors have also been tried
[33,37,38] but these drugs increase the risk of lithium toxicity and may
not be the best choice for additive therapy.
Concluding Remarks
This case report emphasizes the need to balance between concerns
regarding the damage to the kidney which is a result of long-term lithium
treatment to the significant therapeutic benefit of lithium as a mood
stabilizer and anti-suicidal drug. Our patient committed suicide attempts
at two occasions after lithium cessation due to concerns regarding
deterioration in renal function and aggravation of NDI. One of those
suicidal attempts (or others that lithium probably prevented) could “bear
fruit” and kill the patient at an early age. Therefore, treating psychiatrists
and nephrologists must perform a risk-benefit calculation before deciding
to stop lithium in bipolar patients, particularly when the cessation of
lithium is not expected to lead to improvement in kidney function. On
the other hand, it is essential that clinicians pursue preventive strategies
to minimize the risk of developing impairments in kidney function
in lithium-treated patients. This may include: prescribing the lowest
effective dose of lithium; rigorous monitoring of plasma lithium levels in
order to avoid episodes of intoxication and acute nephrotoxicity; annual
examination of creatinine clearance (GFR); avoid co-administration of
other drugs that may increase plasma lithium levels such as NSAIDs, ACE
inhibitors and diuretic drugs; and, assessment of other determinants of
renal function, such as plasma calcium, proteinuria and peripheral edema.
Funding
This research received no specific grant from any funding
agency.
Conflict of Interest
The authors declare that they have no conflict of
interest.
Ethical Considerations
This article does not contain data obtained
from a study with human participants. This type of study did not require
a formal informed consent.
Article Information
Article Type: Case Report
Citation: Shnaider A, Azab AN (2015) Lithium-induced
Nephrogenic Diabetes Insipidus – A Case Report and
Discussion on the Pathophysiological Mechanism.
Int J Nephrol Kidney Failure 1(3): doi http://dx.doi.
org/10.16966/2380-5498.113
Copyright: © 2015 Shnaider A, et al. This is an
open-access article distributed under the terms
of the Creative Commons Attribution License,
which permits unrestricted use, distribution, and
reproduction in any medium, provided the original
author and source are credited.
Publication history:
Received date: 31 August 2015
Accepted date: 9
Sep 2015
Published date: 15 Sep 2015