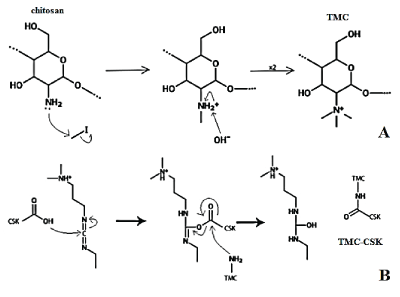
Figure 1: Reaction mechanism for the synthesis of TMC from chitosan (A); Reaction mechanism for the conjugation of TMC and CSK (B).

Guanyu Chen1 Yuan Huang2 Jingyuan Wen1*
1School of Pharmacy, Faculty of Medical and Health Science, University of Auckland, New Zealand*Corresponding author: Jingyuan Wen, School of Pharmacy, Faculty of Medical and Health Science, University of Auckland, 95 Park Road,
Grafton, 1142, Auckland, New Zealand, Tel: +64 9 3737599 Ext: 82762; E-mail: j.wen@auckland.ac.nz
Gemcitabine has shown to be useful in a range of human cancers, but oral delivery of gemcitabine lead to low oral bioavailability due to its high hydrophilicity and low plasma half-life. Therefore, this project developed a peptide CSKSSDYQC (CSK) modified N-trimethyl chitosan (TMC) polymer nanoparticles for oral delivery of gemcitabine, to target intestinal goblet cells thus enhance the drug uptake and its oral bioavailability. TMC was synthesised from deacetylated chitosan using a novel two-step synthesis, then conjugated with CSK. Gemcitabineloaded TMC-CSK nanoparticles were prepared via ionic gelation. Characterisation and cellular drug uptake studies were conducted. Results showed the nanoparticles with average size of 173.6 ± 6.8 nm, PDI of 0.20 ± 0.02 and zeta potential of 18.5 ± 0.2 mV, and entrapment efficiency of 66.44 ± 0.02%. They appear non-spherical shape with minimal aggregation. Drug loaded TMC-CSK and TMC nanoparticles showed sustained drug release manner compared to plain drug solution. For cellular drug uptake, the gemcitabine loaded TMC-CSK NPs have greater cellular drug uptake capability compare to plain drug solution, and demonstrated the cellular uptake process was energy dependent. The study demonstrated the developed TMCCSK nanoparticle is a promising delivery system for oral delivery of gemcitabine.
CSKSSDYQC Peptide; N-trimethyl Chitosan; Oral bioavailability
Gemcitabine (2’-deoxy-2’, 2’ difluorocytidine monohydrochloride [β isomer], dFdC) is an antineoplastic drug effective in treating a variety of malignancies, including pancreatic, breast and non-small cell lung cancers [1]. Orally administered gemcitabine is susceptible to poor intestinal permeation due to its high hydrophilicity, and extensive first-pass metabolism by the enzyme cytidine deaminase, present in both the human gut and liver [2], thus lead to low plasma half-life of approximately 8 minutes [3], and resulting in low oral bioavailability of approximately 10% [4]. These pharmacokinetic factors have the overall effect of limiting bodily exposure to gemcitabine when given via the oral route. Therefore, higher doses of oral gemcitabine are required to reach therapeutic levels, however toxicity in previous trials has forced discontinuation and gemcitabine remains all but restricted to parenteral delivery [5].
Oral formulations are of immense interest as they are thought to be the most acceptable of all dosage forms due to being non-invasive and having high patient preference [4,6,7]. Formulation approaches include co-administration of metabolism inhibitors and modifications to the chemical structure of gemcitabine itself. Co-administration of oral gemcitabine with 3,4,5,6-tetrahydrouridine (a cytidine deaminase inhibitor) has demonstrated modest success, with an oral bioavailability of 40% [8], though other pharmacokinetic aspects were deemed unfavourable. Another study of chemical modification, adding a 3-(dodecyloxycarbonyl) pyrazine-2-carbonyl group to the N4-position on the cytidine ring of gemcitabine, theoretically decreasing deamination and therefore first pass metabolism [9]. This leads to increase half-life, but was only minimally increased, which limits the overall potential of such a delivery formulation [10].
Nanoparticles (NPs) may offer advantages when delivering drugs orally, these are drug carriers within the nanometer range (1- 1000 nm) in which the drug adsorbs on the particle surface, or is encapsulated, entrapped or dissolved in the particle matrix [11,12]. Owing to their small size, NPs have increased ability to permeate through cell membranes, thus allowing for more drugs to reach the systemic circulation [13]. In addition, NPs confer drug protection from degradation and metabolism by the body [14]. The sustained release characteristic of NPs results in reducing the side effects cause by antineoplastic drugs [15,16]. The functionality of NPs is determined strongly by the choice of polymer used in formulation [17]. Chitosan is the second most abundant polymer on earth and is highly biocompatible and biodegradable within human bodies [18]. Chitosan based NPs showed high drug encapsulation and great mucoadhesion in prior research. In addition, it tends to open intercellular tight junctions through interactions with anionic tight junction proteins via cationic molecular groups, therefore enhancing permeability [19]. At physiological pH however, these properties are lost because of deprotonation. Chitosan becomes progressively insoluble as pH is further increased beyond this [20]. N-trimethyl chitosan (TMC) is a partially quaternised chitosan derivative designed to overcome the limitations of chitosan. Unlike chitosan, TMC is soluble at neutral and basic environment which promotes a greater potential anatomical area for NP uptake within the gastrointestinal tract (GIT) [4]. TMC also has superior absorption compared to chitosan through mucoadhesion (thereby increasing contact with intestinal epithelium) and promotion of both transcellular and paracellular absorption [21]. CSKSSDYQC (CSK) is a naturally occurring peptide that actively targets goblet cells, which comprise the second most common group of intestinal epithelial cells [4]. CSK increases the uptake of NPs through caveolae and clathrin mediated endocytosis to occur. This has been shown using HT29-MTX cell and in vivo studies in rats where insulin loaded TMC-CSK NPs resulted in a 1.7 increase in ileal permeation and a 1.5-fold increase in bioavailability compared to TMC NPs [10]. Until now, very few studies of oral delivery of gemcitabine have been reported.
The present study developed a TMC-CSK NP as a novel delivery system for oral delivery of gemcitabine, was investigated for the first time. The gemcitabine loaded TMC-CSK NPs showed promising physical and chemical properties, and had a significant improvement in intestinal epithelial cell uptake compared to drug solution and drug loaded unmodified TMC NPs.
Chitosan >90% deacetylated, MW 400 kDa, was purchased from Comwin Fine Chemicals Co. (Changzhou, China). CSK peptide was purchased from Chinese Peptides Co., Ltd. (Hangzhou, China). Gemcitabine hydrochloride, MW 299.66, iodomethane solution (CH3 I), sodium iodide (NaI), N-(3-dimethylaminopropyl)-N′- ethylcarbodiimide hydrochloride (EDC.HCL), N-methylpyrrolidone (NMP), sodium tripolyphosphate (TPP), ethylenediaminetetraacetic acid disodium (EDTA-2Na), sodium tripolyphosphate (TPP), Polysorbate 80 (Tween 80), 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) were all purchased from Sigma Aldrich (St. Louise, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from Labpartner (Shanghai, China). Dialysis tubing, MW 12,000 was purchased from Membra-Cel® (Viskase, USA). All reagents were of analysis grade. Milli-Q water was supplied by Millipore Millipack40 and filtered through 0.22 µm filter.
One-step TMC synthesis: The reaction process was carried out as depicted. Briefly, 2 g of chitosan and 4.8 g of sodium iodide were dissolved in 80 mL of n-methylpyrrolidone solvent and stirred magnetically for 45 minutes. The reaction was refluxed in the dark over a water bath at 60°C. Fifteen mL of 15% w/v sodium hydroxide solution and 11.5 mL of methyl iodide was added, and stirred for 120 minutes. The product was collected via vacuum filtration and dialysed. The exchange of iodide ions with chloride ions was performed in dialysis process. Briefly, 5% w/v NaCl was used as outer medium for dialysis over 2 days, and subsequently the outer medium was changed to Milli-Q water and further dialysis for 2 days.
Two-step TMC synthesis: The above procedure was carried out identically to the point of product collection. The reaction mixture was instead poured into 200 mL of ethanol and centrifuged at 10,376 g for 10 minutes. The ethanol was removed by washing the mixture with 60 mL of diethyl ether on a glass filter. The filtered product was dissolved with 4.8 g of sodium iodide in 80 mL of n-methylpyrrolidone solvent and magnetically stirred under reflux conditions in the dark, over a water bath at 60°C for 45 minutes. Eleven mL of sodium hydroxide 15% w/v solution and 7 mL of methyl iodide were added and the reaction continued for 60 minutes at 60°C. Two mL of methyl iodide and 0.6 g of sodium hydroxide pellets were then added and the reaction was continued for 60 minutes. After the synthesis was completed, the lyophilized TMC yield was calculated by using the mass of the final harvested TMC polymer over the original mass of chitosan used for synthesis.
Figure 1A shows the TMC methylation step. The spare pair of electrons presents on the primary amine residue of deacetylated glucosamine units of chitosan attacks at the relatively δ+ methyl group of the CH3 I. Simultaneously, the methyl-iodine link is broken via SN2 kinetics. This results in a formation of a secondary ammonium ion with positive charge. Subsequent deprotonation mitigates this charge and forms water, setting up the reaction to repeat. These two steps repeat twice more, thus form an overall positively charged trimethylated chitosan. The use of this novel two-step synthesis method leads to a greater reagent react time, facilitating the likelihood of full amine methylation, therefore forming higher yield of the product [22].
Figure 1: Reaction mechanism for the synthesis of TMC from chitosan (A); Reaction mechanism for the conjugation of TMC and CSK (B).
TMC-CSK conjugation: Following synthesis, 380 mg of EDC.HCL and 228 mg of NHS catalyst were dissolved in 0.6% w/v TMC solution in a nitrogen environment. CSK was added to give a final concentration of 3% w/v, and left to stir at 500 rpm in the dark for 3 days to allow conjugation occurred sufficiently. The resulting TMC-CSK product was collected via dialysis in 2 L of milli-Q water for 3 days. The dialysed product was then freeze dried for 2 days. Figure 1B shows the CSK conjugated with TMC via amide bonds formed between the residual primary amino groups on TMC and the carboxyl groups on CSK [23]. The δ-hydroxyl group of CSK attacks the δ+ carbon atom on the carbodiimide group of EDC. The resulting electron rearrangement and subsequent nucleophilic attack by the amino group in TMC results in cleavage of the ester link in the EDC-CSK complex, and results in amide bond formation between CSK and TMC [23,24].
TMC (quantity shown in table 1) and 50 mg of gemcitabine were dissolved in 5 mL of milli-Q water. Tween 80 (0.07 mL) was added and mixed by magnetically stirring. TPP solution of various concentrations was prepared according to table 1. Two mL of respective TPP solutions were added dropwise and the mixture was stirred for 1 hour at room temperature. All solutions were characterised for particle size and zeta potential, and entrapment efficiency.
| Formulation | [TMC] (mg/mL) |
TMC (mg) |
[TPP] (mg/mL) |
TPP (mg) |
TMC:TPP weight ratio |
| A | 5.00 | 25.0 | 4.17 | 8.33 | 3:1 |
| B | 6.00 | 30.0 | 5.00 | 10.0 | 3:1 |
| C | 7.00 | 35.0 | 5.83 | 11.7 | 3:1 |
| D | 8.00 | 40.0 | 6.67 | 13.3 | 3:1 |
| E | 7.00 | 35.0 | 2.92 | 5.83 | 6:1 |
| F | 8.00 | 40.0 | 3.33 | 6.67 | 6:1 |
| G | 4.00 | 20.0 | 3.33 | 6.67 | 3:1 |
| H | 7.00 | 35.0 | 17.5 | 35 | 1:1 |
Table 1: Formulation parameters for nanoparticle synthesis
Respectively, 25 mg of either chitosan, TMC, or TMC-CSK polymer was dissolved in 5 mL of milli-Q water with 50 mg of gemcitabine. Acetic acid 99.8% (0.1 ml) was added to the chitosan solution to promote dissolution. Tween 80 (0.07 mL) was then added, and the mixture was magnetically stirred at 450 rpm for 5 minutes. As per formulation parameters, 8.33 mg of TPP was dissolved in 2 mL of milli-Q water. This TPP solution was added dropwise to the polymer solution and stirred at 450 rpm at room temperature for 60 minutes.
Particle size, polydispersity index and zeta potential: The particle size, polydispersity index (PDI) and zeta potential of the chitosan, TMC, and TMC-CSK NPs were measured in triplicate with a Malvern Zetasizer Nano-ZS (Malvern Instruments Ltd., Malvern, UK).
Entrapment efficiency: Entrapment efficiency (EE) was determined indirectly using high performance liquid chromatography (HPLC). NPs containing 50 mg of gemcitabine were centrifuged at 42018 g for 10 minutes. The supernatants were filtered and the drug concentrations were determined by HPLC (1260 Infinity, Agilent Corporation, Germany). The EE of the NPs was determined by subtracting the non-entrapped drug from the amount of drug added to the NPs and expressed as a percentage (see Equation 1).
\[EE\% = \frac{{Total{\rm{ }}Drug{\rm{ }}Amount{\rm{ }} - {\rm{ }}Free{\rm{ }}Drug{\rm{ }}Amount}}{{Total{\rm{ }}Drug{\rm{ }}Amount}} \times 100\,\,\,\,\,\,\,\,\,\,\,\,\left( {Equation{\rm{ }}1} \right)\]
Surface morphology: Nanoparticle surface morphology was determined via scanning electron microscope (SEM) (Phillips XL 305 Field Emmsion Gun, Netherlands). Freeze-dried NPs were placed onto metal plates and sputter coated in platinum (Quorum Q150R S rotary pump sputter coater). SEM observations were conducted on TMC NPs and TMC-CSK NPs respectively.
In vitro drug release: In vitro release of gemcitabine from nanoparticle formulations was studied using Franz diffusion apparatus (FDC-6, Logan Instruments, Somerset, NJ). Gemcitabine solution of 1% w/v concentration and an equivalent amount of drug loaded NPs were added to the donor compartment of the Franz diffusion cell, with a cellulose membrane (pore size<100 nm, MW 12,000, Membra-Cel®, USA) sandwiched between the donor and receptor chambers. The receptor chamber was filled with phosphate buffer (pH 7.4), and were kept under constant stirring at 500 rpm and maintained at 37 ± 1°C. Aliquots of 500 µL were withdrawn at pre-determined time points (15 min, 30 min, 1 hr, 2 hr, 3 hr, 4 hr, 6 hr, 8 hr, 12 hr and 24 hr) and replaced with 500 µL fresh pH 7.4 phosphate buffer. Samples were filtered and determined by HPLC.
Goblet cell is the second most frequent cell type on intestinal epithelium which produces mucus on the epithelial wall. The HT29- MTX-E12 cells can be conditioned to acquire the mucus-producing features of goblet cells [25]. Thus, to investigate the goblet cells targeting capability of CSK peptide, HT29-MTX-E12 cells were used for cellular drug uptake studies. HT29-MTX-E12 cells were cultured using Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) containing 10% fetal calf serum, 1% penicillinstreptomycin-glutamine (Life Technology, NY, USA) and 1% nonessential amino acid (Life Technology, NY, USA) at 37°C, 98% humidity and 5% CO2 . Five mL of medium containing HT29-MTX-E12 cells were seeded onto 60 mm culture dish (Corning Coster Corp, USA) at the density of 1 × 105 cells/ml, cellular drug uptake studies were then conducted after the cells reached 90% confluence.
The HT29-MTX-E12 cells were pre-incubated in Hank’s balanced salt solution (HBSS) at 37°C for an hour, and rinsed with HBSS 5 times to remove the secreted mucus on the apical side of the cells before use. Two mL of HBSS transport buffer containing 500 µg/mL drug solution and equivalent drug loaded TMC NPs and drug loaded TMCCSK NPs were added and incubated with the cells at 4°C and 37°C for 2 hours to investigate the effect of incubated temperature on drug uptake. Subsequently, the medium was aspirated off after 2 hours, and rinsed with ice-cold HBSS 3 times. The cells are then scraped off into 1 mL of extraction solution of 0.02 N hydrochloric acid/methanol (1:1, v/v). Acetonitrile/methanol (3:1, v/v) was used as a lysis buffers to lysis the cells, followed by ultrasonication at 60 W for 10 min. The final drug extracted was quantified by HPLC. In addition, the cell protein content was subsequently dissolved in 1 mL of 1 M NaOH, and the determined using abicinchoninic acid (BCA) assay kit (Thermo Scientific, USA). As a result, the amounts of total drug uptake were expressed as the quantity of drug (µg) uptaken per mg cellular protein.
The optimal drug loaded NPs were studied for their physical and chemical stability. The optimal NPs were stored in screw-capped amber vials at three different temperatures (4, 25 and 40°C) for 3 months. Samples were withdrawn at 0, 1, 2 and 3 months and evaluated their physical stability by looking at their particle size and PDI. The chemical stability profile was evaluated by measuring the amount of drug retained in the NPs as drug entrapment efficiency which was described previously
In the control groups, drugs with the equivalent to that entrapped in the NPs were used, and the drug solution was subjected to the 3 temperatures and the drug retained was examined at the end of each month during the 90-day storage.
Statistical data analyses were performed using Microsoft Excel 2010 software (Redmond, WA). Results are shown as mean ± 95% confidence intervals. Data comparisons were conducted using regression analysis, ANOVA tests and two-tailed t-tests. A P-value ≤ 0.05 was pre-established as the minimum level of significance.
From table 2, the one-step synthesis method produced a TMC yield of 43%. By contrast, the two-step method produced a TMC yield of 89%. With regard to nanoparticle formation, formulation G produced a limited yield for evaluation, and was therefore excluded. TMC-CSK conjugation was carried out using 200 mg of TMC to interact with 120 mg of CSK, forming 242 mg of TMC-CSK, which with a yield of 76%.
| Original material and weight | Final material and weight | Percentage yield | |
| One-step Synthesis | 200 mg chitosan | 86 mg TMC | 43% |
| Two-step Synthesis | 200 mg chitosan | 178 mg TMC | 89% |
Table 2: Yield of One-Step Synthesis TMC, Two-Step Synthesis and TMC
The degree of quaternization was determined from the integration of 1H nuclear magnetic resonance (1H NMR, UNITY INOVA-400, Varian Inc., CA, USA). From the results of 1H NMR determination, TMC with a degree of quaternization of 12.7% was obtained; Results of 1H NMR determination had confirmed the TMC and CSK conjugation by the characteristic peaks at 6.752 and 7.023 ppm for two protons of benzene ring of tyrosine in CSK peptide sequence, respectively. In addition, according to the amino acid detection results, the CSK content in TMC-CSK polymer was determined to be 0.09 mmol/g. Moreover, TMC yield from chitosan is functionally dependent upon the degree of chitosan deacetylation. Chitosan deacetylation is the ratio of free amino groups of chitosan not bearing an acetyl group to the total number of nitrogen atoms of chitosan [26,27]. For example, chitosan with a chitosan deacetylation of 75% will have 75% of its nitrogen atoms available for methylation during TMC synthesis [27]. Chitosan with deacetylation >90% was preferred to 75% deacetylated chitosan for formulation purposes, as a higher degree of deacetylation demonstrated a higher TMC yield in preformulation studies. In addition, the one-step methylation method formed comparatively more n-dimethyl chitosan (an impurity), the two-step synthesis led to the formation of TMC with higher quarterisation. Thus, the twostep quarterisation method appears to generate TMC of higher purity and yield compared to one-step synthesis alone. High quarterisation is attractive because it promotes high solubility over a wide pH range [22]. Therefore, the two-step synthesis has been carried out in the current study in a bid to increase methylation at the amine group of chitosan.
From figure 2A, it shows all formulations were in the nanometre range except for formulations D and H, which were sized at 1384.0 ± 113.8 and 1035.5 ± 207.7 nm respectively. Formulations A, C and G had the smallest particle size of 382.9 ± 18.2, 371.9 ± 146.8 and 310.9 ± 5.4 nm respectively. There was an increase in particle size as TMC concentration increased (p=0.004). From figure 2B, only the TMC:TPP ratio of 1:1 was not in the nanometre range as it was sized at 1035.5 ± 207.7 nm. A TMC:TPP ratio of 3:1 had the smallest particle size of 371.9 ± 146.8 nm. Only the difference between 3:1 and 1:1 was statistically confirmed (p=0.007). Figure 2C shows that the particle size of all formulations was in the nanometre range. TMC-CSK NPs exhibited the smallest particle size of 173.6 ± 7.7 nm. Additionally, the particle size of the chitosan NPs, TMC NPs and TMC-CSK NPs were significant different (p ≤ 0.05).
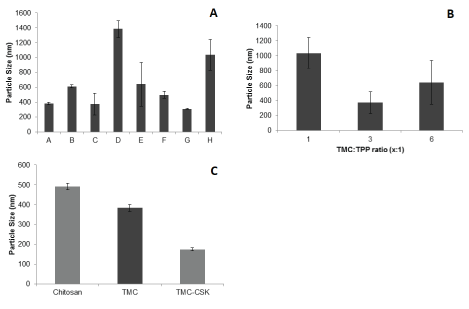
Figure 2: Particle size of TMC nanoparticle formulations A-H. Results are reported as mean ± 95% CI, n=3; B): Particle size of TMC nanoparticle formulations with different TMC:TPP ratios. Results are reported as mean ± 95% CI, n=3; C): Particle size of chitosan, TMC, and TMC-CSK NPs prepared with 4.17 mg/mL TPP and 5 mg/mL of polymer. Results are reported as mean ± 95% CI, n=3.
Comparing NPs prepared by different polymers, TMC NPs displayed a smaller mean size than chitosan NPs (p ≤ 0.05) and is consistent with previous works [28,29]. According to Dehousse et al. [30] the higher charge density on TMC compared to chitosan results in formation of more compact NPs. A theory supported by Ing et al. [28] who claimed that TMC NPs had smaller sizes due to the strong electrostatic interaction with polyanionic TPP [30]. The effect of CSK on reducing particle size is also observed in the present study. This observed difference between TMC-CSK and TMC is statistically significant (t-test p-value ≤ 0.05). This contradicts work by Jin et al. who found that insulin-loaded TMC-CSK NPs exhibited greater size [10]. According to the authors, this may have been due to the introduction of CSK which has a MW of 1018 Da [10,15]. It is however important to note that both insulin and CSK bear an overall negative charge and electrostatic repulsion may have played a role in increasing particle size compared to our formulation. Gemcitabine by comparison of known neutral physiological charge may not have interfered in the electrostatic interactions of TMC and CSK, thereby resulting in more compact NPs compared to insulin.
Particle size is of paramount importance in delivery chemotherapeutic agents because of its implications for permeation and retention [31]. NPs smaller than 300 nm in diameter confer several advantages, including an improved ability to penetrate the mucus layer, increased systemic distribution and enhanced intestinal absorption via M cells and enterocytes [32]. These NPs must be larger than 8 nm, however, as NPs smaller than 8 nm will permeate interendothelial junctions in health tissues leading to reduced targeting and increased side effects [33]. Further, cancer cells have larger interendothelial junctions (between 40 and 1000 nm), which facilitate greater permeation. This, along with poor lymphatic drainage in tumours encourages nanoparticle retention and reduces potential side effects [33]. Win, Feng & colleagues demonstrated that polymeric NPs sized 100-200 nm were optimal for uptake in Caco-2 cell lines [34]. The authors also mentioned that such NPs are believed to be internalised by receptor-mediated endocytosis, while larger ones are more susceptible to phagocytosis [34]. These findings suggest that our TMC-CSK NPs of 173.6 ± 6.8 nm may have gastrointestinal absorptive advantage compared to chitosan and TMC NPs, both with average sizes greater than 300 nm.
PDI is a measure of homogeneity, with a higher PDI implicating a wider size distribution [13]. From figure 3A, it shows formulations A, B, F and G had PDI values less than 0.5 (PDI values of 0.399 ± 0.033, 0.424 ± 0.054, 0.389 ± 0.042 and 0.43 ± 0.027 nm, respectively), suggesting they were more uniform than the rest. This is also reflected in their small 95% CI, implying narrow size distribution and increased homogeneity of the particles. In addition, PDI was observed to increase as TMC concentration increased. Figure 3B shows the PDI values varied by 0.053 across all ratios and all were > 0.5, and the TMC:TPP ratio effect on PDI was not considered to be statistically significant across all TMC:TPP ratio values (p ≥ 0.05). Moreover, from Figure 3C, PDI was < 0.5 for all three formulation groups of Chitosan, TMC and TMC-CSK NPs, while the TMC-CSK NPs showed a lowest PDI of 0.20 ± 0.02. Furthermore, the PDI of the three formulation groups were significant different (p ≤ 0.05).
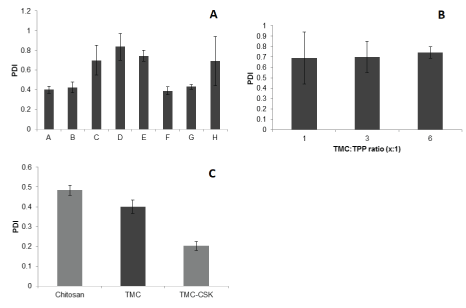
Figure 3: A): PDI of nanoparticle formulations A-H. Results are reported as mean ± 95% CI, n=3; B): PDI of nanoparticle formulations with different TMC:TPP ratios. Results are reported as mean ± 95% CI, n=3; C): PDI of chitosan, TMC and TMC-CSK NPs prepared with 4.17 mg/ mL TP and 5 mg/mL of polymer. Results are reported as mean ± 95%CI, n = 3.
Figure 4A shows all formulations (A-H) were found to have positive zeta-potentials, while the formulations C and E had zeta potentials are of +25.7 ± 1.0 and +12.6 ± 4.6 mV, respectively, which were fallen within the ideal zeta potential range between +10 and +30 mV. Additionally, the zeta potential was observed to decrease as TMC concentration increased. Figure 4B demonstrated the TMC:TPP ratio of 1:1 had a zeta potential of +0.12 ± 0.13 mV, which did not fallen within the ideal range of +10 and +30 mV. While TMC:TPP ratio of 3:1 had the greatest zeta potential of +25.23 ± 1.74 mV, doubling the zeta potential of TMC:TPP ratio of 6:1 and 25-fold greater than that of TMC:TPP ratio of 1:1. Moreover, the zeta potential of the three various ratios groups were significant different from each other (p ≤ 0.05). Figure 4C indicated that only TMC-CSK NPs had a zeta potential of 18.5 ± 0.2 mV, which was within the ideal range of between +10 and +30 mV, and the zeta potential of the chitosan, TMC and TMC-CSK NPs were significant different (p ≤ 0.05).
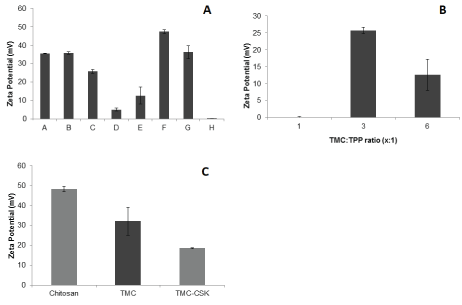
Figure 4: A): Zeta potential of TMC nanoparticle formulations A-H. Results are reported as mean ± 95% CI, n=3; B): Zeta potential of nanoparticle formulations with different TMC:TPP ratios. Results are reported as mean ± 95% CI, n=3; C): Zeta potential of chitosan, TMC, and TMC-CSK NPs prepared with 4.17 mg/mL TPP and 5 mg/mL of polymer. Results are reported as mean ± 95% CI, n=3.
TMC NPs were found to have a lower zeta potential than chitosan NPs (p ≤ 0.05). A high zeta potential is favourable because it represents electrostatic repulsion between particles and thereby confers a degree of formulation stability [35]. TMC NPs typically elicit a greater zeta potential to chitosan NPs. According to Boonyo et al. TMC NPs had higher zeta potential because of the positively charged sites along the TMC chain [29]. Corollary, Sadeghi et al. speculated that the conversion of amino group of chitosan to quaternary ammonium group was responsible for the higher positive charge and zeta potential [36]. TMC synthesis involves the methylation of the primary amine groups present in chitosan, and this methylation itself has been shown to lower the zeta potential of molecules. This is because methyl substitution influences charge distribution in a stabilizing manner [37]. Because of this, the methylation of chitosan might be expected to lower the zeta potential overall. Furthermore, CSK conjugation causes a further fall in zeta potential, which is attributed to the anionic character of CSK. Higher zeta potentials are preferred, however, values greater than 30 mV are unfavourable, since the strongly cationic characteristic can be disruptive to cell membrane integrity [4]. TMC-CSK NPs exhibited a mean zeta-potential of +18.5 ± 0.2 mV, and are therefore less likely to disrupt cellular membranes compared to chitosan and TMC NPs.
Figure 5A indicated that all formulations had EE greater than 70% with an average EE of 73.3%. Formulation D had the highest EE% value of 73.6 ± 0.9%. For chitosan, incorporation of most drugs occurs via interaction with free positive amine groups present in chitosan [4,38]. Gemcitabine interacts through both ionic and hydrogen bonding when formulated via ionic gelation [4]. Since EE is comparable for all polymers, it can be speculated that the interaction still occurs via interactions with residue primary amine groups and available hydrogens, even for methylated TMC (and TMC-CSK). Increased TMC concentration has been speculated to cause greater hindrance to drug entrapment [4]. Therefore, from the results, it was observed that increasing TMC concentration had the effect of increasing EE. Figure 5B demonstrated that all TMC:TPP ratios groups displayed EE values greater than 70%. The highest EE observed was the TMC:TPP of 6:1 ratio with an EE value of 73.4 ± 0.4%. In addition, it was observed that there was no significant different in EE of all TMC:TPP ratios groups (p ≥ 0.05). Figure 5C displayed that all the delivery systems of chitosan, TMC and TMC-CSK NPs had an EE of greater than 65%. TMC-CSK NPs displayed a lower EE than TMC or chitosan NPs by a margin of 0.3%. This difference however, was statistically confirmed (p ≤ 0.05). TMC NPs yielded better EE than chitosan NPs by 0.03% but this difference could not be statistically confirmed (p=0.33).
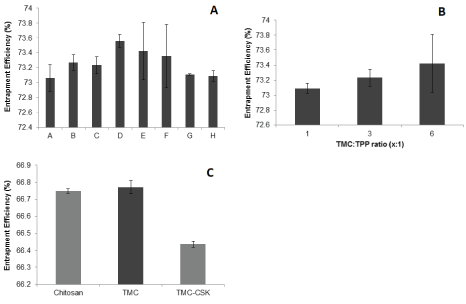
Figure 5: A): EE % values of formulations A-H, each prepared with 50 mg of gemcitabine. Results were reported as mean ± 95% CI, n=3; B): EE% of nanoparticle formulations with different TMC:TPP ratios. Results are reported as mean ± 95% CI, n=3; C): EE% of chitosan, TMC and TMC-CSK NPs, each prepared with 50 mg gemcitabine, 4.17 mg/mL TPP and 5 mg/mL of polymer. Results were reported as mean ± 95% CI, n=3.
When comparing polymers regarding EE, the optimal polymer was TMC, followed by chitosan and TMC-CSK. A t-test comparing the EE of chitosan and TMC showed no statistical difference (p-value of 0.33). TMC NPs had a lower zeta potential compared to chitosan and, due to differences in positive charge density available for ionic interaction by definition, we may have expected to see comparatively less interaction between gemcitabine with TMC compared to chitosan. This proved insignificant however, which may indicate that potential sites of gemcitabine-amine interaction were not saturated under current reaction conditions [2]. TMC-CSK NPs displayed a lower EE value relative to TMC and chitosan NPs. TMC and CSK are large molecules and the conjugation of such molecules creates steric hindrance when a third molecule, such as gemcitabine, attempts to incorporate itself [39]. Moreover, excipients are also known to influence nanoparticle EE. A study by Hosseinzadeh et al. however found no difference in entrapment efficiency when different amounts of gemcitabine was added to chitosan nanoparticle formulations, though considerably smaller concentrations were used (0.2-1.0 mg/mL vs 7.1 mg/mL) [2]. In the future, different concentrations of gemcitabine could be explored to optimise entrapment efficiency of TMC-CSK NPs.
Overall, formulation A exhibited the most favourable combination of particle size of 382.9 ± 18.2 nm, PDI of 0.40 ± 0.03 and zeta-potential of +35.6 ± 0.2 mV. It was thus selected as the model formulation to investigate the differences between chitosan, TMC and TMC-CSK NPs. Formulations D and H were excluded from further characterisation on the basis of size. Formulation G showed favourable characteristics but low yield, indicating that it would not be an economical formulation to manufacture, thus was excluded.
Morphological studies were conducted using SEM. Figure 6 shows the SEM photographs of TMC NPs and TMC-CSK NPs revealed slightly rough surfaces when prepared via ionic gelation, with a polyhedron shape [29,40,41]. It is well established that nanoparticle characteristics are highly dependent on TPP and polymer weight ratio, among other parameters, though this pertains more to nanoparticle size and surface charge rather than morphology or topography [40]. Other research has demonstrated typically spherical polymeric NPs when optimally prepared via ionic gelation because spheres are the most energetically favourable conformation in solution [42]. One theory which may explain our observation has been posed by Wang et al. who postulate that polyhedric shapes are possible where gelation acts to form a nucleus from which polymeric crystallisation occurs, thereby resulting in polyhedral NPs [39]. Additionally, freeze dried NPs are susceptible to expansion, fragmentation, and may elicit more rough surfaces than if otherwise measured as a fresh formulation [4]. The freeze-dried TMC-CSK NPs were visualised as large aggregates under SEM, the nanoparticle suspension was prepared and directly measured by Malvern zetasizer, therefore, less aggregation would be compared to the freeze-dried NPs [40]. Hence the prepared NPs in suspension before freezed-dried may be smaller and more symmetrical than depicted. Moreover, both micrographs show particles within the nano-meter range. Aggregation is minimal across preparations indicating relative nanoparticle stability, which is concordant with our relatively high surface charges determined via electrokinetic studies analysing zeta potential [43].

Figure 6: SEM Micrograph of TMC NPs (A); TMC-CSK NPs (B); and TMC-CSK NPs with more particles (C).
From figure 7, it demonstrated that gemcitabine alone undergoes burst release with 81.6 ± 3.5% of drug release occurring within the first four hours, and 93.7 ± 2.7% within 24 hours. The nanoparticulate delivery forms of gemcitabine demonstrate sustained release of the drug, with a steady diffusion of the entrapped drug form the polymer matrix over 24 hours. The two nanoparticulate delivery systems have similar release profiles. TMC-CSK NPs show relatively slightly more rapid drug release than TMC NPs. At the end of the 24-hour period, 89.3 ± 2.4% of gemcitabine entrapped in TMC-CSK NPs had been released compared to 77.6 ± 4.1% of gemcitabine in TMC NPs.
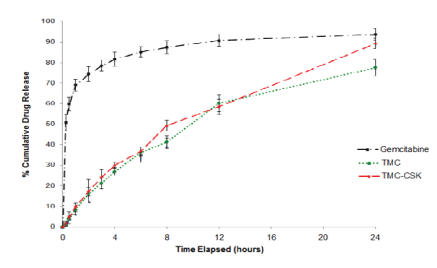
Figure 7: In vitro release of gemcitabine from chitosan, TMC and TMC-CSK NPs in PBS (pH 7.4) at 37°C. Values indicated are mean ± 95% CI, n=3.
Sustained drug release is a desirable parameter of a nanoparticle delivery formulation because it prolongs the effective dose of a given drug [17]. Non-formulated gemcitabine demonstrates rapid release in vitro which has problematic implications, including increased risk of side effects due to cellular exposure in situ, potentially causing local cytotoxic damage and meaning increased frequency of dose administration to sustain a target therapeutic concentration [43]. Conjugated TMC-CSK NPs in turn demonstrated more rapid drug release compared to TMC NPs. This effect may be due to polymeric arrangement at the molecular level, where the large conjugated CSK peptide represents a steric barrier to compact nanoparticle formation, lead to irrespective of the diameter of the nanoparticle itself [10]. This would promote a less dense, more porous nanoparticle which is more readily permeated by water and thus allows a more rapid yet potentially more complete release of encapsulated gemcitabine molecules.
From figure 8, different temperature of 4°C and 37°C were applied to conduct the drug uptake studies. The uptake amounts from gemcitabine solution, gemcitabine loaded TMC NPs, and gemcitabine loaded TMC-CSK NPs after 2 hrs under 37°C were much higher than that under 4°C, suggesting that the uptake was energy dependent. In addition, both gemcitabine loaded TMC NPs and gemcitabine loaded TMC-CSK NPs had significant greater drug uptake amount compared to gemcitabine solution in both 4°C (p < 0.05) and 37°C (p < 0.01), and gemcitabine loaded TMC-CSK NPs have greater drug uptake amount compared to gemcitabine loaded TMC NPs (p<0.01). Moreover, the experiment performed at 37°C showed 1.9-fold greater uptake than that at 4°C for drug loaded TMC NPs. While for drug loaded TMCCSK NPs, there was a 2.3-fold greater uptake at 37°C than at 4°C.
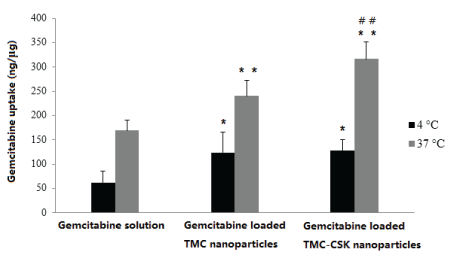
Figure 8: Temperature-dependent uptake of 500 µg/mL of gemcitabine solution, gemcitabine loaded TMC NPs, and gemcitabine loaded TMC-CSK NPs after 2 hr at 4°C and 37°C (mean ± SD, n=3; Significant difference from control, *: p<0.05, **: p<0.01; Significant difference from TMC NPs, ##: p<0.01).
Endocytosis, an energy dependent process, is blocked at low temperature [45]. Upon incubation at 37°C, the cells are metabolically active and energy consuming uptake can occur. In contrast, at 4°C, the metabolism is reduced so that binding of NPs to the cell membrane is reduced. Our results provided evidence that both the TMC NPs and TMC-CSK NPs cellular uptake are mediated by endocytosis. This is in accordance with a previous research which demonstrated that endocytosis is the main mechanism of TMC based NPs uptake by HT29-MTX-E12 cells [4]. In addition, the presence of CSK peptide promotes cellular uptake was due to the capability of CSK to target intestinal goblet cells. Moreover, the drug loaded TMC-CSK NPs have a remarkable increase in cellular uptake compared with drug solution over 2-hour period. Note that 2 hours is sufficient for the gemcitabine loaded NPs to reach small intestine and achieve maximal intestinal epithelial cellular uptake, which indicates great benefit for intestinal drug absorption. Therefore, in this cellular uptake study, TMC-CSK NPs demonstrate great benefit for intestinal drug uptake, thus lead enhance the drug oral bioavailability. Furthermore, although from previous drug release study, gemcitabine release from TMC-CSK NPs is faster than TMC NPs, it remains to be seen the advantages that CSK offers to cellular drug uptake into systemic circulation, since this is an important factor for future clinical use.
The optimized TMC NPs and TMC-CSK NPs were studied for their stability at different conditions and time intervals. Over the 90- day storage period, the optimal formulations were withdrawn and evaluated for size, and retained drug amount. The results of physical and chemical stability of the two optimal nanoparticulate formulations are shown in tables 3 and 4.
As seen from the particle size analysis data (Table 3), the particle size of the NPs was stable at 4°C for up to 3 months, for 2 months at 25°C conditions. The results revealed that storage temperature had a noticeable effect on the stability. At higher temperatures, the NPs showed quicker aggregation than at cold temperature. This might be due the thermal energy imparted to vesicles causing both the rate and force of collision between vesicles to increase. The collision and aggregation led to the increased particle size. These findings suggest that the storage temperature of choice of 4°C for prepared NPs showed minimal changes in particle sizes over 3 months.
| Batch # | Storage conditions | Particle size (d.nm) | |||
| 0 Month | 1 Month | 2 Months | 3 Months | ||
| Drug loaded TMC NPs | 4°C | 382.9 ± 18.2 | 388.9 ± 28.2 | 382.9 ± 8.2 | 392.9 ± 11.4 |
| 25°C | 382.9 ± 18.2 | 392.9 ± 24.1 | 402.1 ± 24.2 | 407.9 ± 22.0 | |
| 40°C, 75% RH | 382.9 ± 18.2 | 402.1 ± 17.0 | 441.5 ± 19.1 | 452.9 ± 27.8 | |
| Drug loaded TMC-CSK NPs | 4°C | 173.6 ± 6.8 | 168.6 ± 11.2 | 183.6 ± 2.9 | 178.6 ± 12.1 |
| 25°C | 173.6 ± 6.8 | 175.6 ± 16.4 | 183.0 ± 7.7 | 198.5 ± 4.5 | |
| 40°C, 75% RH | 173.6 ± 6.8 | 201.4 ± 15.4 | 244.5 ± 26.1 | 257.2 ± 13.3 | |
Table 3: Influence of storage conditions on the particle size of lyophilized optimal NPs (mean ± SD, n = 3)
Another stability parameter to determine is the drug leakage from the prepared NPs over the 90-day storage period. The percentages of drugs retained in NPs were measured for three months at the three different temperatures: 4, 25 and 40°C. The effects of storage temperatures on drug amount retained in the NPs over the 90-day period are presented in table 4. The table shows the initial drug content of the optimal NPs analysed gradually reducing over time, indicating the temperature has significant effect on the particle stability. Particularly, at 40°C/75% RH, a relatively significant change was observed in the drug content for both TMC NPs and TMC-CSK NPs over 3 months storage. Drug content was reduced to approximate 83% and 74% for TMC NPs and TMC-CSK NPs, respectively in the third month. Reduce of drug content at high temperature and moisture may be caused by the polymer degradation at such conditions [46].
| Batch # | Storage conditions | Amount of gemcitabine (% of initial amount) | |||
| 0 Month | 1 Month | 2 Month | 3 Month | ||
| Drug loaded TMC NPs | 4°C | 100 | 102.1 ± 4.2 | 97.1 ± 2.2 | 103.1 ± 2.0 |
| 25°C | 100 | 93.4 ± 6.2 | 86.1 ± 3.7 | 87.4 ± 6.6 | |
| 40°C, 75% RH | 100 | 85.7 ± 8.5 | 91.1 ± 2.2 | 83.1 ± 4.8 | |
| Drug loaded TMC-CSK NPs | 4°C | 100 | 99.2 ± 5.2 | 98.1 ± 3.4 | 91.4 ± 2.6 |
| 25°C | 100 | 87.4 ± 5.3 | 92.1 ± 5.1 | 90.3 ± 2.6 | |
| 40°C, 75% RH | 100 | 83.7 ± 8.5 | 78.1 ± 12.2 | 74.1 ± 11.8 | |
Table 4: Influence of storage conditions on the chemical stability of gemcitabine on lyophilized optimal NPs (Mean ± SD, n = 3)
Based on the observation, the drug loaded TMC NPs and TMCCSK NPs should be stored at 4°C, where they remained stable in terms of both particle size and drug content.
Gemcitabine is an anticancer drug with an established clinical role. Its poor oral bioavailability however has limited it to parenteral delivery only. The development of new drug delivery systems is to allow for effective oral gemcitabine delivery. In this study, TMC was synthesised from chitosan via a novel two-step methylation method, then conjugated with CSK and formulated to NPs. The TMC-CSK NPs showed relatively smaller particle size, and were within optimal zeta potential range, as well as promising EE for oral drug delivery. Both TMC NPs and TMC-CSK NPs displayed sustained drug release manner. The TMC-CSK NPs show significant improvement in cellular uptake compared with drug solution and TMC NPs, demonstrating the goblet cells targeting capability of CSK peptide. Based on the stability studies, the drug loaded TMC NPs and TMC-CSK NPs should be stored at 4°C, where they remained stable in terms of both particle size and drug content. The results of our research suggest that TMC-CSK NPs confer favourable properties for oral drug delivery, and can be used as a preliminary groundwork for future studies with the ultimate aim of creating an oral drug delivery system of gemcitabine that can used in both a clinical and community setting.
Pharmacy 410 group students’ contribution in this research project. They are Angus Tan, Alanna Adams, Pooja Kumar, Ryan Moxham Smith, and Georgina Soo.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Chen G, Huang Y, Wen J (2018) Development of an Advanced Delivery System of CSKSSDYQC Peptide Modified N-Trimethyl Chitosan Nanoparticles for Oral Delivery of Gemcitabine. J Med Chem Drug Des 1(2): dx.doi. org/10.16966/2578-9589.109
Copyright: © 2018 Chen G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access