Figure 1: Structure of Cholesterol.

Betsy Reshma G1 MSA Muthukumar Nadar1 P Mosae Selvakumar2*
1Department of Biosciences and Technology, Karunya University, Coimbatore, Tamil Nadu, India*Corresponding author: P Mosae Selvakumar, Department of Chemistry, Karunya University, Coimbatore 641 114, Tamil Nadu, India, E-mail: mosae@karunya.edu
Cholesterol is the integral part of cell membrane which forms a major reason for cardiovascular diseases, a manifestation of hypercholesterolemia. Cardiovascular disease is the leading cause of death worldwide. The failure to eliminate plasma cholesterol is due to the imbalance in production of apolipoprotein B by the liver and clearance of Low Density Lipoprotein (LDL) by LDL receptors. Transport of cholesterol in our body by the five classes of lipoproteins potentially explains hypercholesterolemia. A loud and clear understanding of the disease leads to the contest of anti-cholesterol drugs. Therapies were arrived by identifying an enzyme or receptor involved in biosynthesis pathway and developing an inhibitor which interferes with cholesterol synthesis. When it comes to the treatment of high cholesterol statins assume the centre stage. Some of the explored drugs and their mechanism are discussed in the current review which will harbinger new grounds for effective delivery of drug and drug design.
Cholesterol; LDL; Hyper-cholesterol drugs; Mechanisms; Atherosclerosis
Cholesterol is present in tissues and also in plasma. The increased level of cholesterol in plasma attribute to hypercholesterolemia. Hypercholesterolemia increases the risk of cardiovascular diseases. Population of people with raised cholesterol level report accounts for 2.6 million deaths and 29.7 million disabilities. In 2008 global prevalence of raised cholesterol among adults was (>5mmol/l) which results in 30 per cent. The conditions manifested by high-cholesterol include atherosclerosis, coronary heart disease, stroke and peripheral vascular disease accounts for one third of all deaths in middle and older adults [1]. The lipid hypothesis in atherosclerosis and other cardiovascular diseases has been powerfully endorsed. Though atherosclerosis is a multifactorial disease it is plausible to target hyperlipidaemia. To design drug, the cholesterol biosynthesis pathway, the rate limiting steps and the degradation pathway is delineated. The therapies emerge by targeting enzymes prime in cholesterol biosynthesis. HMG-CoA reductase the rate limiting enzyme for cholesterol biosynthesis is targeted at expression, transcription, posttranscription and degradation levels. HMG-CoA (3-hydroxy3-methylglutaryl-CoA) gets converted to mevalonic acid by HMG-CoA reductase which is the precursor of isoprenoids. Lipitor has made over $125 billion in sales which is about quarter of Pfizer’s revenue [2]. With enormous market space for anti-cholesterol the demand for new targets should be sorted. This review exhibit existing drugs and therapies along with their detailed mechanism and targets (Figure 1).
Figure 1: Structure of Cholesterol.
Cholesterol is present in rapid equilibrium in plasma [3]. The transport of cholesterol is well studied. Cholesterol is a lipophilic molecule which is transported by lipoproteins. The buoyant density and Apo lipoprotein composition is the determining factor of the type of lipoprotein. The forward transport of cholesterol is initiated by the secretion of low density chylomicrons (~100 nm) from dietary fats. In blood chylomicrons acquire apoE and apoC11. The chylomicrons is taken up by the hepatocytes is reassembled to VLDL (Very low density lipoprotein) followed by IDL (Intermediate density lipoprotein) and terminally to LDL (Low density lipoprotein) [4,5]. The reverse transport is the essential process of cholesterol transport. HDL (High density lipoprotein) collects the free cholesterol from the peripheral cell by cell surface receptors. Cholesterol from blood is transported back to liver in by reverse transport. The amphipathic cholesterol gets absorbed into HDL. As HDL gets matured to be alpha HDL it processed by direct and indirect pathway. The direct pathway is achieved by Scavenger receptor B1 [6]. The indirect pathway involves the conversion of HDL to VLDL, IDL and LDL. In response to increase in LDL in blood the hepatocytes express LDL receptors for receptor mediated endocytosis of LDL [7].
Biosynthesis of cholesterol is initiated by three step conversion of acetyl-CoA to 3-hydroxy-3-methylglutaryl Co-A (HMG-CoA) (Figure 2). HMG-CoA is reduces to mevalonate by HMG-CoA reductase, is the rate limiting step in biosynthesis pathway. Series of phosphorylation reaction activate and decarboxylate mevalonate to isopentenyl pyrophosphate. The rearrangement to allylic pyrophosphate yields prenyl pyrophosphate by condensation. Two molecules of farnesyl pyrophosphate condense head to head produces presqualene pyrophosphate. 2,3-Oxidosqualene is the result of epoxidation which circularize to lanosterol. Lanosterol reduction yield cholesterol [8]. Figure 2 explains the importance of biosynthesis pathway and related enzymes in formulating potential drugs.
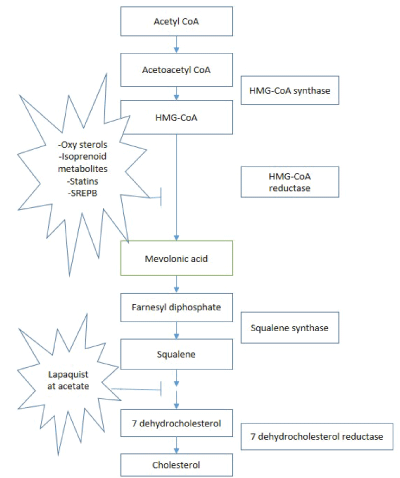
Figure 2: Biosynthesis pathways of cholesterol and drug targets.
The overview of drug action and its mechanism with drugs shown in Figure 3. The fibrates induce lipoprotein lipolysis by peroxisome proliferation. The response is from the transcription factors known as peroxisome proliferation activator complex receptor (PPARs). PPARs heteromerize with retinoid receptor binds to response element in the DNA. Fibrates are synthetic ligands to the PPARs but specifically target PPAR alpha. The hypotriglyceridemic action of fibrates induces the expression of LPL (Lipoprotein lipase) but repress apo-CIII. This will enhance the LPL mediated catabolism of VLDL particles [9]. Hyperlipidaemic patients have increased level of plasma Tumour necrosis factor alpha and interferon gamma. The fact is LDL stimulate T lymphocytes and macrophages to release IF gamma and TNF alpha. Fibrates though not very superior treatment method effectively decrease lipids and cytokines [10]. The common fibrates Clofibrate and Ciprofibrate cause hepatomegaly. Gemifibrozil and Fenofibrate are hit medications in the family of fibrates they are known for milder peroxisome proliferation [11]. Dosage requirement of Lipofen-150 mg/day.
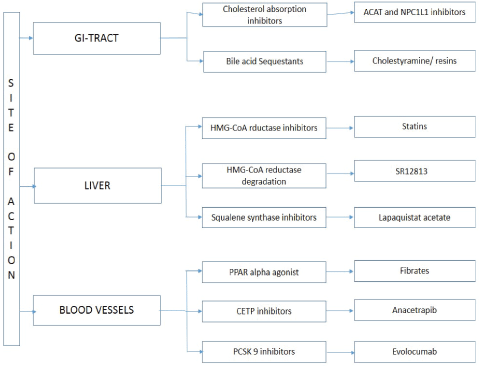
Figure 3: Different target mechanisms of anti-cholesterol drugs.
There are two ways which contribute to plasma cholesterol, one is absorption from diet (300-500 mg) and other is endogenous biosynthesis of cholesterol. The cholesterol esters get converted into free cholesterol by cholesterol ester hydrolases before entering the cell. The sequential step is the conversion of cholesterol esters to free cholesterol esterol by Acetyl Co-A cholesterol acyltransferase (ACAT), which is packed in chylomicrons and enter the plasma. The debate was whether ACAT or cholesterol ester hydrolase will be the better target. It was proved that ACAT inhibitor is effective in reducing the absorption of cholesterol rather than Cholesterol ester hydrolase because dietary bulk is in free form [12]. The potent ACAT inhibitors are Avasimibe has undergone animal studies helps in plaque rupture by clearing out cholesterol esters [13].
NPC1L1 Inhibitor
Niemann-Pick C1 like 1protein (NPC1L1) is an important site of interstinal cholesterol uptake. Enterocytes absorb cholesterol which is packed into chylomicrons as cholesterol esters is taken up by hepatocytes. By blocking NPC1L1 receptor ezetimibe decreases cholesterol absorption. Ezetimibe in combination with statins effectively reduce hepatic cholesterol pool [14].
Bile Acid Sequestrants
Cholestyramine and colestipol influences the hepatic metabolism. Cholesterol 7 alpha hydroxylase is the rate limiting step in bile acid synthesis. Composition of bile acid: cholic acid 30-40 per cent; Chenodeoxycholic acid 30-40 per cent, Deoxycholic acid 20-25 per cent and lithocholic acid 1-2 per cent. The idea is to consume bile acids which encourage the cholesterol to bile acid metabolism in hepatocytes. The new demand created by bile acid depletion will increase HMGCoA reductase and LDLR expression several folds thereby reducing plasma cholesterol level [15]. The bile acids in fixed amount circulate in bowels and liver called the enterohepatic circulation. The bile acid synthesis is stimulated when 20 per cent of bile acid is diverted [16].
The catabolism of cholesterol is initiated when bile acids are captured by anion exchange resins. The bile acids are evacuated by anion exchangers. The natural response is the activation of cholesterol 7 alpha hydrolases for obvious reason that cholesterol is the precursor of bile acids. The LDL receptors in the hepatic cells pop up and LDL cholesterol in the blood is recovered for biosynthesis of bile acids. Hence the cholesterol level is drastically lowered [17]. Colesevelam is the new line of drug. It works by attaching to food substance. It is passed out without any absorption by the intestine.
LDLR Expression
Berberine: Berberine is an alkaloid isolated from Huanglian (Coplischinesis) a Chinese herb. Found to show the highest expression of LDLR expression. The art behind the action is to articulate the LDLR and LDL cholesterol homeostasis. The high expression of LDLR in hepatic cells improves the clearance of LDL cholesterol in the plasma by receptor mediated endocytosis [18]. The intracellular cholesterol regulates LDLR expression by negative feedback mechanism. Sterol regulatory element (SRE1) in the promoter region of LDLR gene is triggered by Sterol regulatory element binding protein (SREBP) coupled with SREBP cleavage activation protein (SCAP). The cholesterol depletion in normal conditions is sensed by the cholesterol sensing domain of the SCAP which aid in the transport of SREBP from Endoplasmic reticulum to the Golgi bodies. In the Golgi apparatus the appropriate cleavage mechanism is carried out. Then SREBP bind to SRE-1 thereby increasing the expression of LDLR. The role of berberine is to increase the expression of LDLR independent of the intracellular cholesterol. The transcription factors of cholesterol mediated feedback regulation will not be involved in berberine action [19].
HMG- CoA Reductase Degradation: SR12813 (tetra ethyl 2-(3, 5 di-tert-butryl 4- hydroxyl-phenyl) ethenyl-1, 1- bisphosphonate), the mechanism of this synthetic drug is to block the endogenous cholesterol biosynthesis by rapid degradation of HMG-CoA reductase. The HMG-CoA reductase can be targeted at different levels. The expression can be supressed, the transcription can be blocked which is RNA centric by (oxy) sterols [20], the post transcription can be regulated to truncate the enzyme by isoprenoid metabolites [21]. SR12813 is targeting the HMG-CoA reductase by degradation which is direct suppression of the cholesterol biosynthesis. The HMG-CoA degradation brought forth by steroid accelerated degradation. The product of mevalonate apart from cholesterol includes other steroid and non -steroid compounds. Accumulation of steroids will block SCAP- SREBP complex by retarding them in the ER thereby blocking cholesterol synthesis and uptake. The sterols induce the binding of SCAP to ER retention proteins Insig-1 and Insig-2 [22,23]. The reductase enzyme also coincides with the sterol induced binding of Insig. Because of the accumulation of the ubiquitinated forms of the enzyme it becomes obligated to destruction by proteasome [24]. The end product and non-sterols of mevolonate combine to accelerate the degradation of reductase by ubiquitin proteasome pathway [25]. SR12813 work independent of the mevolanate derived metabolites, the degradation is rapid by a mechanism that primes the enzyme for degradation [26].
Inhibition of HMG-CoA Reductase: The family of statins are the most hit drugs in the market that served the heat for nearly four decades. The mechanism is directly blocking the HMG-CoA reductase. When the intracellular cholesterol decreases the LDLR receptor expression is enhanced in order to capture the LDL cholesterol that is vagrant in the plasma [27]. Statins also tend to inhibit hepatic synthesis of apolipoprotein B100 and decrease the synthesis and secretion of triglyceride rich lipoproteins [28]. Some statins are atorvastatin, lovastatin, rosuvastatin, simvastatin, pitavastatin, fluvastatin. Simvastatin takes its place in WHO Model List of Essential Medicine.
Squalene synthase inhibitors: Squalene synthase catalyse the reductive dimerization of farnesyl di-phosphate to squalene the precursor of cholesterol. The search for squalene synthase inhibitors have been intensified because of the unique location in the pathway. The screening of natural compounds, synthetic compounds and rational designs were carried out [29]. In spite of the widespread success of statins it contributes to myotoxicity. Squalene inhibitors are proven to increase the expression of LDLR without inhibiting the production of other intermediates [30]. Statins along with squalene synthase inhibitors show protective effect on statin induced myotoxicity. They also continue to be excellent lipid lowering drugs complementing statin [31]. Lapaquistat acetate reached phase III clinical trials, unfortunately withdrawn because of hepatotoxicity [32]. Stilbenoid derived small molecules that can up regulate the LDLR.
PCSK-9 Inhibitors: The boon of monoclonal antibodies and the treatment of hyperlipidaemia has shown greater propensity. FDA approved Alirocumab (Praluent, Safoni) in July 2015, the new class of drugs known as proprotein convertase subtilisin/kexin type 9 (PCSK9) [33]. In August FDA approved evolocumab (Repatha, Amgen) the second in PCSK9 inhibitors. PCSK9 is a protein involved in regulating LDLR recycling [34,35]. Evolocumab is fully human monoclonal antibody that binds to PCSK9 which retards the interaction with LDLR [36].
Apolipoprotein inhibitors: To intensify the cholesterol lowering potential alternative therapies were under investigation. Apolipoprotein B100 is a mandatory component of LDL cholesterol and other atherogenic lipoproteins. Assembly of apoB containing lipoproteins takes place in the endoplasmic reticulum with the aid of microsomal triglycerides [37]. Direct inhibition of apoB sounds brilliant but is not plausible yet. Mipomersen is a modified oligonucleotide which is complementary to the human apoB mRNA sequence [38]. When mipomersen hybridize with mRNA it results in the activation of RNase H which catalyses the cleavage of RNA eventually inhibiting the protein translation to decrease apoB concentration. More appropriate in the treatment of Familial hypercholesterolemia. In January FDA approved mipomersen as an orphan drug.
CETP inhibitors: Cholesteryl ester transfer protein (CETP) promotes transfer of cholestyeryl esters from HDLs to other lipoprotein. By inhibiting CETP increase HDL-C eventually decreasing atherogenic non-HDL particles [39]. Genetic defects in CETP showed elevated HDL-C. When humans were given CETP inhibitor drugs HDL-C and apoA-1concentration increased as a bonus in some cases decreased LDL-C and apoB [40]. The atheroprotective role of CETP inhibitors is manifested by HDLs. 1) Promote efflux of cholesterol from macrophages. 2) Inhibit oxidation of LDLs. 3) Inhibition of vascular inflammation and thrombosis [41]. Anacetrapib inhibits CETP by tight binding of CETP to HDL thereby making it unavailable to shuttle cholesteryl esters [42] (Table 1).
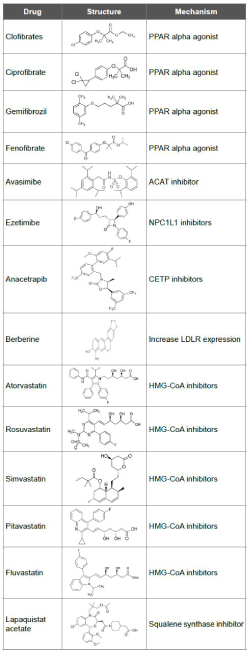
Table 1: Structure and mechanism of anti-cholesterol drugs.
Quantitative Structure-Activity Relationship is performed to determine the pharmacophore requirement that pull out the activity. Also it suggests the favourable and unfavourable interactions around the molecule. 3D-QSAR studies are carried out for drug-ligand binding studies. The application of force field renders significance to the activity of the molecule. Lennard-Jones potential is used for superimposing drug crystallography data with trained set of data of known function. Comparative molecular field analysis (CoMFA) determines the steric and electrostatic fields which are correlated by partial least square regression. 3D QSAR studies on CETP inhibitors proposed hydrophobic fields are the major contributors followed by electrostatic, hydrogen bond donor and steric fields [43]. Studies on QSAR modelling for HMG-CoA reductase inhibitors were carried out in order to narrow down the structural relationship to activity in the diversity of molecules available. With the knowledge of chiral topological indices QSAR modelling predicted HMG-CoA reductase inhibition activity of new chiral isomers. It was evaluated that pyrimidine and pyrrole substitution resulted in partial inhibition of HMGCoA reductase [44]. Table 1 represents the drug structure and their mechanism. The structure and activity relation is important when it comes to fabricating a potential drug.
The review adumbrated the various enzymes and mechanism of cholesterol biosynthesis and transport. The possible classes of mechanism enable the decision of choosing the right medication and the concomitant pairs. New grounds are open to improve drug delivery systems of effective drugs. QSAR studies lead to the investigation of the structure that is responsible for its effect. It holds immense ability in modelling precise drugs, with high efficiency. With the understanding of the mechanism of drugs available in market, cost effective and potential benefit drugs can be designed and proposed. Risk assessment is an important part in clinical decision. Statins have been the mainstay in LDL lowering drugs. Antisense RNA and monoclonal antibodies are the new line of treatment. New interventions focus on favourable benefitrisk ratio.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Reshma GB, Nadar MSAM, Selvakumar PM (2018) A Review on Anti-Cholesterol Drugs and their Mechanisms. J Med Chem Drug Des 1(1): dx.doi.org/10.16966/2578-9589.104
Copyright: © 2018 Reshma GB, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access