
Figure 1: Chemical vs bio catalyzed synthesis of sitagliptin.

Andrés R Alcántara*
Department of Chemistry in Pharmaceutical Sciences, Faculty of Pharmacy, Complutense University of Madrid, Madrid, Spain*Corresponding author: Andrés R Alcántara, Department of Chemistry in Pharmaceutical Sciences, Faculty of Pharmacy, Complutense University of Madrid, Plaza de Ramon y Cajal, s/n. E-28040 Madrid, Spain, Tel: 00-34-913941820; E-mail: andalcan@ucm.es
The use of biocatalysts in chemical syntheses is becoming very significant in Green Chemistry; in fact, synthetic routes mediated by enzymes or cells are generally conducted under mild reaction conditions, at ambient temperature and can use water as reaction medium in many cases. The exquisite enzymatic precision (in terms of chemo-, regio- and stereoselectivity) avoids the need of functional group activation and protection/deprotection steps, usually required in traditional organic synthesis. Furthermore, modern techniques of genetic manipulation are creating enzymes even more precise and robust for being used in industrial processes. Accordingly, biocatalysis allows procedures which are shorter, produce less waste and reduce manufacturing costs and environmental impact. These features are even more significant in drug synthesis; a crucial area inside Medicinal Chemistry, since it is well known Pharma Industry is beyond a doubt the most wasteproducer. Thus, in this article we will comment some archetypical biocatalyzed protocols for the preparation of bioactive molecule and/or chiral building blocks for drug synthesis, to illustrate the great advantages of introducing biotransformations in synthetic pathways.
Biocatalysis; Biotransformations; Drug synthesis; Green chemistry; Enzymatic Precision
Biotransformations can be defined as the use of natural biocatalysts– enzymes– for the transformation of non-natural manmade organic compounds [1]. Definition of biocatalysis is as simple as the use of enzymes in chemical synthesis [2]. Both terms are used to describe the application of enzymes, either as isolated preparations or in whole cell format (both in their native cells or as recombinantly expressed proteins in alternate host cells) in a given synthetic schedule, with the aim of taking advantage of the exquisite enzymatic precision inherent to their use, in terms of chemoselectivity (selective transformation of one functional group in the presence of others), regioselectivity (formation of a regioisomer over another one) or stereoselectivity (formation of one stereoisomer over another) [3]. The use of biotransformations has increased considerably over the last few decades, complementing classical chemical synthesis in multiple industries, including pharmaceuticals, fine chemicals, or food [4-18]. On the other hand, and based on the principles and metrics of green chemistry and sustainable development, biocatalysis is absolutely a green and sustainable technology, as recently reviewed by Sheldon and Woodley [18]. To support this affirmation, these authors point out the following considerations:
Hence, biocatalyzed procedures not only are cheaper and generate less waste than conventional organic syntheses, but also are more environmentally attractive, more costeffective, and therefore more sustainable [19]. Then again, bio transformations with isolated enzymes can be carried out in standard multipurpose batch reactors, thus eluding the need for very expensive specific devices, such as high-pressure equipment [20,21]. Last but not least, because enzymatic processes are generally conducted under approximately the same conditions of temperature and pressure, multiple transformations can be easily coupled in cascade processes, therefore resulting in a very economically and environmentally attractive methodology [2,22-24].
On the other hand, it is well known that pharma industry is by far the most waste-producer, as observed upon calculation of the Environmental factor– mass of waste/mass of product, usually expressed as kg/kg, for assessing the environmental impact of manufacturing processes of active pharmaceutical ingredients (APIs) [25], this value is higher than 100, and can be nearly 200 in pre-clinical stages, indicating the huge amount of residues generated. For this reason, it is not surprising to check how bio catalysis is being increasingly implemented in pharma industry involved in drug synthesis [2,12,16,26-41,42-44].
Nevertheless, this increase has not been linear, being rather modest until the last two decades, when a clear burst has been observed [2], this explosion has been caused by the genetic manipulation of biocatalysts by directed evolution [45-51], what has been named the “third wave in biocatalysis” [5]. In fact, although enzymes are very active and selective biocatalysts, for industrial application, a very common reason to engineer them is to increase their stability under reaction conditions, because those ones needed for a biocatalytic procedure may differ dramatically from those present in a cell, therefore demanding high temperatures, extremes of pH, high substrate and/or product concentrations, oxidants, and organic co-solvents. Sometimes an enzyme must tolerate these conditions for only a few minutes or hours, but in a continuous manufacturing process, an enzyme may need to tolerate them for months. There are many ways to increase robustness of biocatalysts, being their immobilization probably one of the most traditionally studied and used [52-58]. But, as commented before, the implementation of directed evolution, that is, the rapid generation of enzyme mutants using new molecular biology techniques, combined with selective pressure via screening conditions [2], has enormously contributed to the preparation of new biocatalysts, able to work efficiently in experimental conditions very different from the “natural” ones, in terms of temperature, pH, presence of organic solvents, etc., while keeping or even increasing its exquisite precision. Anyhow, as pointed out by Truppo [2], we are still just scratching the surface of potential biocatalytic applications, because the next phase (“4th wave”, biocatalysts engineered through rational directed evolution in a design−make−test cycle combining multiple disciplines in one seamless industrialized workflow) will start when we were able to increase (between 2-10 times) the rate of the overall process.
To illustrate this “need for speed” we would exemplify some archetypical cases of the employ of a biocatalyst in drug synthesis.
Probably, the most representative example is the sitagliptin process used in the commercial manufacture of antidiabetic Januvia®, from Merck, the first marketed oral antihyperglycemic drug belonging to the gliptin family [59]. Sitagliptin can be used either alone or combined with metformin or thiazolidinedione, another oral antihyperglycemic agents in the treatment of Type 2 diabetes mellitus. Sitagliptin is the most widely sold dipeptidyl peptidase-4 (DPP-4) inhibitor in the US and worldwide, reaching sales of 6,358 million USD in 2014 with an expected rise to 7,525 in 2020. Sitagliptin was the second leading antidiabetic product in 2014, after insulin glargine, and is predicted to be the leading product by 2020 [60,61].
The first chemical synthesis of sitagliptin [62] involved asymmetric hydrogenation of an enamine 2 chemically derived from pro-sitagliptin 1 using a rhodium-based chiral catalyst (Rh [Josiphos]), at high pressure (Figure 1); nevertheless, this process is not stereoselective enough (97% ee), and the final product is contaminated with rhodium, so that different additional purification steps are required. Some other chemical syntheses have been recently reviewed by Davies et al. [63].
Figure 1: Chemical vs bio catalyzed synthesis of sitagliptin.
Nevertheless, an enzymatic process has substantially improved the efficiency of sitagliptin manufacturing [64,65]; in fact, using an engineered transaminase, developed at Codexis by rational design, a biocatalyst with broad applicability toward the synthesis of chiral amines was obtained. Thus, an (R)- selective transaminase (ATA-117, a homolog of an enzyme from Arthrobacter sp.), was used as stating point, and it was modified to accommodate non-naturally recognized bulk substrates as prositagliptin 1, initially by a rational design through structural homology model of ATA-117, using a substrate walking approach with atruncated methyl ketone analog of 1. Accordingly, the large binding pocket of the enzyme was first engineered, and then evolving that enzyme for activity toward 1, leading to a better variant containing 12 mutations. Anyhow, as this engineered enzyme was not yet of practical utility, further evolution rounds (rounds 3 to 11) focused on increasing enzyme activity and in-process stability [66]. Finally, under optimal conditions, the best variant converted 200 g/L pro-sitagliptin ketone 1 (Figure 1) to sitagliptin 3 with a 92% yield and an enantiomeric excess higher that 99%, by using 6 g/L enzyme in 50% DMSO. The biocatalytic process provides sitagliptin with a 10-13% increase in overall yield compared to the chemical process, a 53% increase in productivity (kg/L per day), a 19% reduction in total waste, the elimination of all heavy metals, and a reduction in total manufacturing cost. Furthermore, the enzymatic reaction is run in multipurpose vessels, so that specialized high pressure hydrogenation equipment is no longer needed. Full details of this process, which obtained the Presidential Green Chemistry Challenge Award (Greener Reaction Conditions Award) from the U.S. Environmental Protection Agency (EPA) in 2010 (http://www. epa.gov/greenchemistry/pubs/pgcc/past.html), can be found in literature [44,66-68,]. Since this innovative approach of biocatalyzed synthesis of sitagliptin using transaminases, other similar examples have been described [69,70].
Coming back to the Merck process, the directed evolution of the transaminase took one year, and subsequently the enzymatic transamination required a refile with regulatory agencies, as the product was already on the market by the time the biocatalytic synthesis was ready for implementation [2]. In this particular example, a doubling in speed should have been enough for fulfilling Merck’s needs, but, as commented before, it would be highly desirable to reach a 10-fold increase in the speed of the overall biocatalyst optimization.
Coming back to the Merck process, the directed evolution of the transaminase took one year, and subsequently the enzymatic transamination required a refile with regulatory agencies, as the product was already on the market by the time the biocatalytic synthesis was ready for implementation [2]. In this particular example, a doubling in speed should have been enough for fulfilling Merck’s needs, but, as commented before, it would be highly desirable to reach a 10-fold increase in the speed of the overall biocatalyst optimization.
Montelukast 7 (Figure 2) is an orally active selective leukotriene receptor antagonist used for the treatment of asthma, originally developed by Merck under the name of Singulair®. The synthetic route [71,72] requires large amounts (to produce annual volume of approximately 20 metric tons per year) of solvents and (−)-β-chlorodiisopinocampheylborane [(−)-DIPchloride], a chiral reducing agent, toxic, corrosive and moisture sensitive, causing burns if it is allowed to contact the skin; furthermore, the chemical reduction of ketone 5 must be carried out at −20 to −25°C to achieve the best stereoselectivity, and requires 80 stoichiometric amounts of [(−)-DIPchloride], so that its replacing should eliminate a major source of waste and cost in the process.
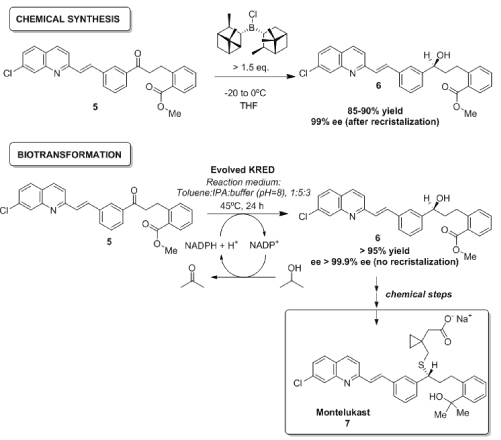
Figure 2: Chemical vs bio catalyzed synthesis of montelukast.
An enzymatic alternative for reducing starting ketone 5 was created by researchers at Codexis and Arch Pharm Labs Limited, which developed a ketoreductase (KRED) by directed evolution, using high-throughput screens mimicking the actual process conditions [73]. Beneficial mutations obtained during each round were recombined and new mutations were introduced, guided by ProSAR, with the target of finding a biocatalyst not more stereoselective than starting KREDs (very stereoslective, leading to ee>99.9%), but rather to efficiently convert substrates as different as bulky ketone 5 and small isopropanol showing higher activity than the initial bioreduction tests (0.1-0.2 g/L of alcohol 6 after 24 h, target ≥ 100 g/L in ≤ 24 h) [73]. The productivity of the final enzyme was improved 2000-fold and stability was also substantially increased, thus allowing a multikilogram process furnishing 233 kg of intermediate 6 (97.2% yield, >99.9% ee), which is not soluble in the reaction medium, therefore shifting the equilibrium in a very effective manner and avoiding the necessity of any further recrystallization. Regarding the cofactor regeneration, the engineered KRED was also capable to oxidize isopropanol (auxiliary substrate) very efficiently, not requiring any other enzyme for this purpose, therefore avoiding the time-consuming evolution of another biocatalyst. Recently, some Chinese companies have developed some modifications of the bioreduction [74,75], while other chemical catalysts have been also tested [76].
Synthesis of the lateral chain of Atorvastatin
Another representative example is the multiton-scale Codexis protocol for the preparation of ethyl (R)-4- cyano-3-hydroxybutyrate (Figure 3, HN10), also known as “hydroxynitrile” (HN), key molecule for furnishing the lateral chain of atorvastatin 11 and other statins: because of the great overall demand of HN required for atorvastin synthesis (estimated to be in excess of 100 metric tons [77], it is highly desirable to reduce wastes and hazards involved in its manufacture while reducing its cost and maintaining or, preferably, improving its quality. In fact, the nucleophilic substitution of ethyl (S)-4-chloro-3-hydroxybutanoate (S-CHBE, 9) with cyanide, the classical chemical protocol, generates a huge amount of residues (E>100), so a three-enzyme two-step process, depicted in Figure 3, increase the quality and sustainability of the purely chemical synthesis.

Figure 3: Codexis’s three-enzyme two-step process for the synthesis of HN 10.
Hence, the first step involves the biocatalytic reduction of 8, using a genetically improved keto reductase (KRED) in combination with a modified NADP-dependent glucose dehydrogenase (GDH) for cofactor regeneration, leading to 9 in 96% isolated yield and >99.5% ee. In the second step, a halohydrin dehalogenase (HHDH), an enzyme capable of catalysing the elimination of halides from vicinal haloalcohols, resulting in epoxide ring formation [78], also catalytically enhanced by directed evolution, was employed to catalyse the nucleophilic substitution of chloride by cyanide. The efficiency and greenness of this protocol (Codexis was awarded the U.S. Environmental Protection Agency’s Presidential Green Chemistry Challenge Award in 2006 for this work [79] is based on the fact that all previous manufacturing routes to HN involved, as the final step, a standard but troublesome SN2 substitution of halide with cyanide ion in alkaline solution (pH=10) at high temperatures (80ºC), being this reaction substituted in the Codexis protocol [40]. In fact, in the SN2 chlorine substitution, both 9 and 10 are base-sensitive molecules, and extensive by-product formation is observed, leading to high E values [77]. Moreover, the product is a high-boiling oil, and a troublesome high-vacuum fractional distillation is required to recover 10, resulting in further yield losses and waste, and clearly contravening the 1st and 6th principles of Green Chemistry [80]. Thus, conducting the cyanation reaction under milder conditions at neutral pH, by employing the enzyme, HHDH, is the key step for increasing the greenness of the overall process.
Regarding the enzymes used in this methodology, both the wild-type KRED and GDH, as well as HHDH, initially displayed very low activities, so that in the first experiments, huge enzyme loadings were required to obtain an economically feasible reaction rate, therefore leading to troublesome emulsions, which hampered the subsequent downstream processing; additionally, although wild-type enzymes were highly stereoselective indeed, they showed severe product inhibition and poor stability under operating conditions [81], and therefore they should be improved to enable a practical large-scale process. Thus, the three enzymes were optimized by in vitro enzyme evolution using gene shuffling technologies according to predefined criteria and process parameters, resulting in an overall process in which the volumetric productivity per mass catalyst load of the cyanation process was improved ~2500-fold, comprising a 14-fold reduction in reaction time, a 7-fold increase in substrate loading, a 25-fold reduction in enzyme use, and a 50% improvement in isolated yield [77].
Many other cases could be commented to illustrate the great potential for the (industrial) use of biocatalysts in drug synthesis, as covered in the increasingly number of revisions published in literature, and previously mentioned in this manuscript. This is far from my purpose; I just want to draw attention to this green and powerful tool, which is here to complement organic chemistry, and to increase the sustainability of the whole process of drug synthesis. I would like to end by complementing Truppo’s masterful prediction on the future of biocatalysis [2]: the future is not only bright, but rather bright green.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Alcántara AR (2017) Biotransformations in Drug Synthesis: A Green and Powerful Tool for Medicinal Chemistry. J Med Chem Drug Des 1(1): dx.doi.org/10.16966/2578-9589.102
Copyright: © 2017 Alcántara AR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access