Introduction
It has long been recognized that acute alcohol intoxication can cause death from accidents or violence and that long-term misuse can increase the incidence of certain kinds of cancer and multi-organ diseases affecting the heart and the liver. According to a 2014 report prepared by the World Health Organization, Global Status Report on Alcohol and Health, an alcohol attributable death occurs every 10 seconds. Yet, increasing evidence suggests that regular consumption of light-to-moderate amounts of alcohol (i.e., one to one and one half standard units) can reduce the risk of Ischemic Heart Disease (IHD) and Sudden Cardiac Death (SCD) [1-6]. These same studies uniformly reported that higher doses of chronic alcohol consumption (>3 standard units) cancel the beneficial cardiac effects of the lower doses of alcohol, promoting Coronary Artery Disease (CAD), SCD, cardiomyopathy, hypertension, and heart failure [7,8]. Cardiac and multi-organ diseases induced by chronic higher doses (>3 units) of alcohol consumption result in an average of 22 years of reduction in life expectancy compared to the general population [9]. According to the National Institute on Alcohol Abuse and Alcoholism, one U.S. “standard” drink contains ~15 grams of pure alcohol, which is found in 12 ounces of regular beer (~5% per volume alcohol), in five ounces of wine (~12% per volume alcohol) and 1.5 ounces of spirits (~40% alcohol). Therefore, up to 15 g/day is considered “light-to-moderate,” and greater than 30 g/day is considered “high” [3,4].
The potential benefits of light-to-moderate doses of alcohol consumption, however, has not received universal acceptance. A recent systematic review of epidemiological data by The Global Burden of Disease Study by Alcohol Collaborators challenged the health benefits of light-to-moderate levels of alcohol consumption. This meta-analysis concluded that the level of alcohol consumption that minimized harm “across all health outcomes particularly the risk of cancer, was zero” [10]. This seeming controversy stems from the fact that certain diseases like Atrial Fibrillation (AF), stroke, [11] hypertension, heart failure, cardiomyopathy, cancer, and liver disease do not manifest any benefit from alcohol, [5,12,13] while other diseases do. The potential mechanisms of the observed benefits of low-to-moderate alcohol consumption in CAD and SCD remain undefined because the observed epidemiologic data are associative and not causative, providing no mechanistic insight by which alcohol causes it’s beneficial or its ill effects. Since the questions that motivate most studies in the health sciences are not associational but causal in nature, it is important to elucidate the possible underlying biological processes involved for preventive and therapeutic purposes. Whenever possible, such knowledge can be acquired by clinical studies and through more aggressive investigations using animal models of human disease. Here, we provide potential mechanisms of CAD and SCD and discuss in some details the U-shaped response of SCD to increasing doses of alcohol.
Epidemiological studies on SCD and CAD
In a landmark study in 1994, Doll and Associates determined the incidence of all-cause mortality in relation to the average amount of pure alcohol consumed per day. The study was performed on 12,000 male middle and older aged British doctors, and was followed-up over 13 years. Those who reported drinking an average of 20 g/day had significantly lower all-cause mortality than those who consumed no alcohol. However, when the consumption became greater than 30 g/day, all-cause mortality increased. In other words, too little or too much alcohol intake was associated with a higher risk of death than moderate intake of alcohol. These opposing effects of alcohol produced a U-shaped relationship between the relative risk of all-cause mortality and the amount of alcohol consumed [3]. The figure 1 illustrates schematically the U-shaped relationship between the relative risk of mortality and the dose of alcohol based on multiple epidemiological studies. This large cohort study confirmed an earlier observation made on a smaller cohort involving 152 subjects of both sexes. This study suggested that 1-3 standard drinks per day reduced the risk of death caused by sudden pulselessness [1]. The results of these earlier studies are consistent with recent meta-analysis studies demonstrating selective benefits of low-to-moderate alcohol consumption in reducing the relative risk of CAD that causes ischemic heart disease [14] and SCD [4]. Furthermore, a meta-analysis, conducted across both genders, that included more than 84 studies, demonstrated a selective benefit in drinkers of <60 g/day alcohol compared to non drinkers, [15] consistent with previous studies [1,3,14]. However, alcohol’s cardioprotective effect on IHD mortality was lost when alcohol consumption was above 60 g/day [15].
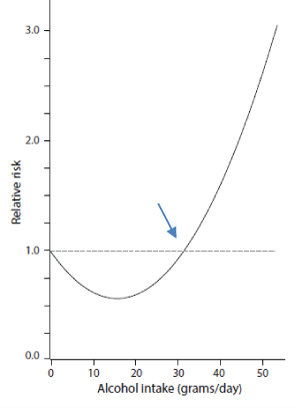
Figure 1: Schematic U-shaped curve representation of relative risk of sudden cardiac death relative to alcohol intake observed in large epidemiological studies. Notice that consumption of >30 g/ day (arrow) raises the relative risk as uniformly shown in multiple independent epidemiological studies in both genders.
The mechanisms by which low doses of alcohol reduce the risk of myocardial ischemia remain elusive. While the antioxidant effects of red wine (The French Paradox) [16] was proposed as a potential mechanism in preventing CAD, it is becoming clear that it is actually the dose of pure alcohol and not the source (e.g., wine, beer, liquor) that provides risk reduction of cardiac disease [4]. However, not everyone agrees with this as “non-alcoholic components of alcoholic beverages” (i.e., polyphenols) may also play a role in the protective effects of alcoholic beverages on health. Multiple factors may contribute to alcohol’s protective effect against CAD. The two major consequences of alcohol’s modulation of these risk factors are: reduction in the risk of blood clot formation and prevention of intravascular plaque development and rupture. Alcohol’s effects on these risk factors include, but are not necessarily limited to, the activation of the fibrinolytic system, lowering of platelet aggregation, and improved endothelial function [1,17]. Importantly, lowering LDL cholesterol (bad cholesterol) [18] and elevating HDL cholesterol (good cholesterol) while reducing C-reactive protein levels greatly diminish the risk of CAD [18,19]. It is thought that these effects of alcohol on blood constituents and improving the lipid profile reduce the risk of atherosclerosis by preventing plaque buildup in the coronary arteries and preventing the formation of blood clots, reducing the risk of myocardial ischemia [6,19]. Interestingly, a recent study found that red wine consumption increases coronary artery calcification which increases plaque stability, significantly reducing the risk of myocardial ischemia and infarction compared to non-drinkers [19]. Whether or not this effect is related to pure alcohol or the antioxidant constituents of red wine remains to be defined.
While epidemiological data on pooled dose-response data based on 28 cohort studies have shown an association between heavy alcohol consumption and increased risk of IHD, the causal mechanisms of such an association are complex and multifactorial [20]. No randomized controlled clinical trial has tried to establish and unravel the mechanism of increased risk of IHD caused by CAD in heavy smokers. Such trials would be extremely difficult to conduct given the almost obligatory presence of confounding risk factors such as smoking, hypertension, and misdiagnosis of cardiomyopathy as CAD [8]. It must be noted that prolonged chronic heavy drinking can result in the most extreme form of cardiac tissue damage, cardiomyopathy [21].
Potential mechanism of U-shaper response of SCD
Interestingly, patients specifically stratified by risk of SCD manifest a U-shaped relationship between the dose of alcohol and the risk of SCD. The mechanism of SCD, i.e., death occurring within one hour of symptom onset (unconsciousness) [4] is most often caused by Ventricular Fibrillation (VF), [22-26] a rapid, disorganized activation of the ventricles [27-30]. A potential confounding factor of alcoholmediated VF may result from the fact that alcohol also promotes AF, which in turn increases the risk of VF. Some 35% to 62% of instances of Paroxysmal AF (PAF) presented to an emergency room are alcoholrelated, with onset frequently 12 to 36 hours after cessation of binge drinking (Holiday Heart Syndrome) [11]. The NHLBI’s Cardiovascular Health Study cohort study showed that the incidence rate of SCD is higher in patients with AF (2.9 per 1000 per year) compared with non-AF controls (1.3 per 1000 per year) [31]. Today an estimated 2.7- 6.1 million people in the United States have AF, affecting more than 33.5 million people worldwide AF. With the aging of the U.S. and world population, this number is expected to increase [31]. Since the relationship between AF risk and the dose of alcohol is linear, [11] it seems clear that the recent observation of a U-shaped relationship between the dose of alcohol and the risk of SCD must result from trigger mechanisms other than AF. A large prospective cohort study involving 85,067 women from the Nurses’ Health Study (currently known as the Women’s Health Study) evaluated the relationship between the dose of alcohol consumed and the risk of SCD in the absence of AF [4]. The study, which began in 1976 and continued until 2004, was conducted on women aged 30-55 years during entry into the study and who were free of chronic diseases. The SCD cases in this study were separated from the non-SCD cases and were documented by medical records and through reports from next of kin [4]. During this study period, SCD occurred in 295 cases, non-sudden coronary heart disease deaths in 987 cases, and nonfatal Myocardial Infarction (MI) in 2,195 cases [4]. The relative risk of alcohol in causing VF (SCD) in this study was found to be U-Shaped and was adjusted for other major risk factors known to directly or indirectly affect the relative risk of VF. These risk factors included coronary heart disease, hypertension, diabetes, body mass index, parental history of MI, smoking, age, and physical activity. Chiuve, et al. [4] identified a U-shaped association between alcohol intake and risk of SCD. Specifically, they fit a Cox proportional hazards model to estimate the relative risk of SCD for various levels of alcohol consumption and controlling for various covariates, like smoking, hypertension, diabetes [4]. To test for the U-shaped relation, they fit a model of the form:\[\lambda \left[ {\frac{{t{\rm{ }}}}{{X,Y}}} \right] = {\lambda _0}\left( t \right){e^{\alpha Y}} + \beta {X^2}\] where X is a continuous measure of alcohol consumption and X represent other covariates such as smoking, age, etc. The X2 term is what lends the model its U (curvilinear) shape. This model was found to be statistically significant (p-value of the coefficient term β was <0.02 adjusted for different sets of covariates). Interestingly, when the analogous coefficient incorporated X instead of X2, the model exhibited a linear relationship and most importantly was not statistically significant. While this may suffer recall bias (the participants were asked to recall how often they consumed alcohol over the past), nevertheless given the relatively large number of participants the demonstration of U-shaped association remains valid [4]. Among women who consumed 5 to 15 g/day of alcohol, the relative risk of SCD was significantly reduced compared to abstainers. With this respect, the relative risk of SCD dropped by 36 percent when women consumed between one-half to one drink per day [4]. However, the relative risk of SCD increased when consumption exceeded 30 g/day. These results are consistent with earlier studies in males and in subjects of both genders [3,32]. Interestingly, alcohol’s beneficial effect on SCD was independent of its source, e.g., beer vs wine vs spirits, [4] suggesting additional factors providing benefit other than the antioxidants present in red wine proposed in the French Paradox [16]. Presently, there is no study that provides insight into the mechanism(s) of alcohol’s pro- and anti-VF effect. It then becomes clear that an understanding of the mechanisms of a U-shaped association between the level of alcohol and the relative risk of SCD is based on defining the mechanism(s) by which alcohol exerts its anti-VF effect at low doses but pro-VF effect at high doses.
Potential mechanisms of VF
VF is characterized by multiple wavelets and constantly forming foci caused by triggered activity [23,33,34]. Here, we propose possible electrophysiological and biochemical signaling mechanisms of alcohol’s dose-dependent pro-VF and anti-VF effect. Microscopic and macroscopic cardiac tissue structural discontinuities profoundly modulate cardiac depolarizing wave conduction, promoting Unidirectional Conduction Block (UCB), re-entrant excitation, and increased risk of VF [35-37]. Multiple factors cause tissue discontinuities, like muscular trabeculae (small muscular bridges connecting large tissue mass) and increased aging and disease-induced cardiac interstitial fibrosis [30,38,39]. Discontinuous propagation and UCB occur when propagation proceeds through a narrow strand and suddenly encounters a large mass of tissue. Under these conditions, a mismatch between the upstream depolarizing current “source” in the narrow strand and the downstream repolarizing current “sink” (i.e., large mass of tissue at rest) develops preventing the wave to propagate from the narrow strands to a larger tissue bulk. However, impulses can propagate from the larger tissue mass (large source) to the narrow strands (small sink) as demonstrated in patterned monolayers of cardiac myocytes, [40-43] and in isolated cardiac tissues [35-37] and in explanted human hearts with cardiomyopathy [44]. Electrical propagation at the very site of source-to-sink mismatch (i.e., the site of sudden tissue expansion) is greatly affected by the degree of local cell-to-cell coupling. Partial uncoupling only minimally affects the upstream source in the narrow strands of muscles but greatly diminishes the downstream sink effect caused by increased tissue impedance [43]. This asymmetrical effect is termed “paradoxical” because of the greater influence of partial cellular uncoupling on the sink rather than the source, an effect that suppresses UCB and restores bidirectional conduction patterned tissue culture [40,43] and in simulation studies [45]. Further increases in cellular uncoupling promote bidirectional conduction block, wavebreaks, and formation of multiple wavelets [28]. These findings indicate partial uncoupling restores normal conduction at sites with UCB while further increases in degrees of uncoupling promote bidirectional conduction block and wavebreak [40,43]. It then follows that drug-induced partial cellular uncoupling imparts non-symmetrical effects on the direction of impulse propagation in discontinuous cardiac tissue [43]. Aliphatic alcohols such as heptanol, [46] ethanol, [47] and palmitoleic acid [40] cause partial cellular uncoupling at low doses and complete uncoupling at higher doses, with ethanol being the least potent cellular uncoupler [47]. Low doses of aliphatic alcohols slightly slow conduction velocity due to partial uncoupling without affecting the source, i.e., fast Na current both in atria [46] and in ventricular tissue [48]. Wavebreaks with higher doses of ethanol promote the formation of multiple wavelets [47] simulating fibrillation-like state [49]. These experimental findings raise the hypothesis that low concentrations of alcohol, by inducing partial cellular uncoupling, eliminates UCB and protects the heart against reentrant VT/VF, while higher doses cause conduction block (wavebreaks) leading to VF. Higher doses may of alcohol also produce VF by prolonging the QT interval in humans increasing the dispersion of repolarization and the risk of torsade-depoints (VT) and VF [50,51]. Electrophysiological studies in humans have shown that intravenous alcohol administration in heavy chronic drinkers promotes electrically inducible VT/VF [51]. It is highly likely that the interaction between dispersion of repolarization and discontinuous propagation act synergistically to promote reentry and VF at macroscopic size scale [41]. Higher doses of ethanol and its metabolite the acetaldehyde reduce inward rectifier potassium current (IK1) [52,53]. Reduced repolarization facilitates the emergence of after potentials [45] and the risk of VT/VF. In addition, higher doses of chronic administration of alcohol in mice alter the distribution of gap junctional Ca43 proteins and increase the structural discontinuities of ventricular tissue. Under these conditions, the vulnerability to VF increases as evidenced by the increased susceptibility to electrically inducible VT/VF in these mice [54].
Potential molecular pathways of VF
Preliminary evidence suggests that the neurophysiological response to low-to-moderate doses of alcohol [55] may reduce the risk of VF by reducing the levels of stress and anxiety through decreasing the sympathetic outflow to the heart. Increased sympathetic outflow to the heart increases the risk of SCD [56-58] and IHD [6,59]. The improvement of overall feelings and performance with low dose alcohol was suggested be mediated, at least in part, by the calming and/or sedative effects of the alcohol [60]. However, not all studies subscribed to this scenario. For example, in alcoholics, alcohol ingestion does not appear in general to relieve anxiety and may increase anxiety in susceptible subjects during a drinking binge. Clearly, more research is needed to assess the validity of the anxiety-reducing theory for alcohol abuse [6]. These findings suggest that low dose alcohol as a stress reducing agent requires individualized assessment of each subject by the caring physician as it cannot be prescribed as a “drug” to reduce tension across different individuals [61]. Advice to consume any amount of alcohol must balance the potential benefits against potential risks related to other chronic diseases.
Finally, in addition to our hypothesized electrophysiological mechanisms, activation of the stress enzyme c-Jun N-Terminal Kinase (JNK) [62] phosphorylates and activates the arrhythmogenic cardiac Ca-calmodulin-dependent protein kinase II (CaMKII) (“kinase on kinase”) [63]. Activation of the highly arrhythmogenic CaMKII [64] increases the late inward Na and Ca currents, promoting afterpotentials and rapid triggered activity [65] leading to VT/ VF [30,66,67] and AF [67,68]. By increasing cardiac sympathetic activity, high doses of alcohol increase the “leakiness” of Ca2+ from the Sarcoplasmic Reticulum (SR) via the ryanodine receptors promoting calcium waves, early and delayed afterdepolarizations (EAD & DADs respectively) that initiate rapid triggered activity leading to AF and VF [68-72].
Conclusions
This mini-review is based on a large number of prior epidemiological studies showing light-to-moderate doses of alcohol (ethanol) consumption reduce the risk of SCD while abstinence or higher doses increase the risk of SCD caused by VF. Based on alcohol’s ability to cause dose-dependent cellular electrical uncoupling, we postulated that lowto-moderate doses of alcohol prevent Unidirectional Block (UCB), reentry and VF while very little or no alcohol consumption preserve UCB and the vulnerability to reentry and VF. In contrast, higher doses of alcohol increases cellular uncoupling promoting conduction block, reentry and VF. Multiple additional mechanisms are also involved in promoting VF with toxic doses of alcohol including activation of the arrhythmogenic enzyme CaMKII. Recommendation of alcohol use to reduce the risk of SCD must however, be strictly individualized by the caring physician and not be contraindicated by other factors including atrial fibrillation. Here, it seems pertinent to conclude with a humorous quote from the founder of evolutionary biology, Charles Darwin, who stated, “An American monkey, after getting drunk on brandy, would never touch it again, and thus is much wiser than most men.”
