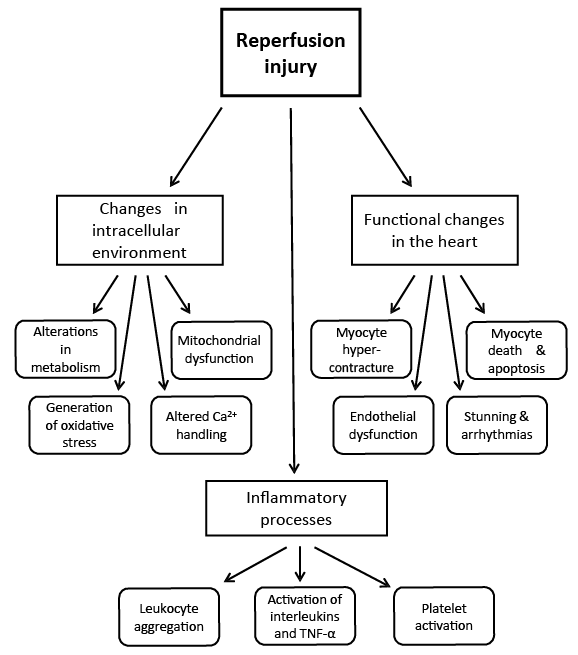
Figure 1: Schematic design describing the consequences of I/R injury in the heart.

Annie Ducas1 Monika Bartekova2 Naranjan S Dhalla1*
1Institute of Cardiovascular Sciences, St. Boniface Hospital Research Center, Canada*Corresponding author: Naranjan S Dhalla, Institute of Cardiovascular Sciences, St. Boniface Hospital Research Centre, 351 Tache Avenue, Winnipeg, Canada, Tel: 204-235-3417; Fax: 204-233-0347; E-mail: nsdhalla@sbrc.ca
Ischemia Reperfusion (I/R) injury is a consequence of reperfusion of the ischemic myocardium when reperfusion is carried out beyond a certain time period of the ischemic insult. The I/R injury is associated with impaired heart function as well as myocardial cell damage and is generally seen to occur during coronary angioplasty, cardiac by-pass surgery, cardiac transplantation and thrombolytic therapy. Several mechanisms including the occurrence of oxidative stress, activation of inflammatory processes, development of intracellular Ca2+- overload, depletion of high energy stores and increased activities of proteolytic enzymes have been suggested to explain the I/R-induced cardiac dysfunction. While contractile failure, apoptosis and necrosis in the heart may be due to cationic redistribution and metabolic alterations as a consequence of oxidative stress and intracellular Ca2+- overload, marked alterations in cardiac gene expression and translation mechanisms may play a critical role in attenuating the recovery of ischemic myocardium. This article therefore is focused on understanding changes in the metabolic and molecular processes occurring in the heart due to I/R injury. Furthermore, current and potential pharmacologic as well as non-pharmacologic interventions are indicated for preventing the I/R injury in the heart.
Ischemia-reperfusion injury; Myocardial cell damage; Cardiac inflammation; Oxidative stress; Intracellular Ca2+- overload
Reperfusion of the ischemic myocardium by Percutaneous Coronary Interventions (PCIs) such as coronary bypass surgery, angioplasty and thrombolytic therapy is essential for the improvement in morbidity and mortality for patients who suffer from ischemic heart disease [1-3]. However, if the reperfusion is not instituted within a certain timeperiod of the ischemic insult, several cardiac complications including arrhythmias, impaired recovery of cardiac function and myocardial cell damage become evident [4,5]. Such abnormalities, defined as IschemiaReperfusion (I/R) injury, are also seen during cardiac transplantation. Thus, a great deal of research has been carried out to understand the mechanisms of I/R injury as well as to develop different interventions which may improve clinical outcome of patients with ischemic heart disease. Indeed, there have been considerable improvements in understanding the pathophysiologic processes underlying the I/R injury and multiple targets for the development of drug therapy have been identified [3,6-8]. In this regard, several preclinical studies have shown a great promise for different agents to exert beneficial effects on I/R injury; however, when these interventions were tested in clinical trials, the results were disappointing. Perhaps the failure of clinical trials with several cardioprotective agents may be related to the fact that the clinical investigations were concerned with the treatment of patients with I/R injury whereas the preclinical studies were carried out to determine the preventive effects of different drugs. Furthermore, the pre-clinical studies seem to be designed for preventing the impact of myocardial ischemia on the reperfusion-induced defects in the heart. It is therefore of critical importance to understand the pathophysiology of myocardial ischemia to gain insight into the mechanisms of reperfusion injury to the ischemic heart. This review is focused on the current knowledge of the pathophysiologic processes due to I/R injury with respect to changes in the metabolic processes, development of oxidative stress, generation of inflammation as well as structural damage to subcellular organelles. This article is also intended to describe the functional effects of I/R injury to emphasize its clinical manifestations. Attempts are also made to explore the therapeutic value of pharmacologic agents including inhibitors of cation channels and exchangers, antiplatelet agents, vasodilators, anti-inflammatory agents as well as protease inhibitors from the prevention and treatment viewpoints. In addition, the cardioprotection afforded by interventions such as pre-conditioning, post-ischemic conditioning and remote ischemic conditioning against I/R-induced abnormalities are discussed.
After an ischemic insult to the heart, a complex series of cellular metabolic changes occur that set the layout for the generation of I/R injury. The beginning of this is the switch from aerobic to anaerobic metabolism in the ischemic myocardium. Cessation of aerobic metabolism causes the loss of high-energy phosphates stores such as ATP and creatine phosphate in cardiomyocytes; the depletion of energy stores is associated with a shift to anaerobic glycolysis [6,9]. It is pointed out that anaerobic metabolism leads to decreased intracellular pH and changes in the cation distribution within cells. This metabolic shift due to myocardial ischemia also generates excess lactate within cardiomyocytes increasing the osmotic load in the cellular environment and creates ultrastructural changes including cell swelling as well as mitochondrial, sarcoplasmic reticulum and sarcolemmal abnormalities [10]. The activity of mitochondrial Pyruvate Dehydrogenase (PDH) remains depressed up to 30 minutes after reperfusion [11,12]. Since the post ischemic recovery of contractile dysfunction in the heart is dependent on PDH activity [12], this metabolic defect is a potential target for the development of cardioprotective therapies.
As perfusion to the myocardium is re-established, there is a large shift of intracellular and mitochondrial Ca2+ ions from the Sarcoplasmic Reticulum (SR) as a result of alterations in the sarcolemmal ion pumps [6,13]. The process for altered calcium metabolism starts when accumulation of excess protons in cardiomyocytes during anaerobic metabolism triggers the activation of sarcolemmal Na+ -H+ ion exchanger as a mean to re-establish ionic gradients [14]. This ion transporter removes H+ ions in exchange for Na+ ions leading to extremely high concentration of sodium within the cell. The intracellular hypernatremia activates the sarcolemmal Na+ -Ca2+ exchanger allowing entry of Ca2+ into the cell and removing the excess Na+. Elevation of intracellular Ca 2+ is also caused by the failure of Na+ -K+ ATPase to remove Na+ ions. This defect in Na+ -K+ ATPase appears to be secondary to changes in the intracellular environment, specifically lowering the pH and elevating the Ca2+ concentration. The Na+ -K+ ATPase activity is also depressed by the depletion of high-energy phosphates during the anaerobic metabolism [13] as the development of intracellular Ca 2+ overload will deplete ATP by stimulating different Ca2+ ATPase activities [6]. These conditions cause structural changes to the transmembrane pumps and exchangers, and affect amino acid as well as sulfhydryl group ends of proteins, and thus attenuating their cation translocation activities.
The next step during the development of I/R injury is dysfunction of the mitochondria. As stated previously, the mitochondria are damaged in the reperfusion process by changes in the osmolarity, pH and shifts of Ca 2+. With re-establishment of blood flow, channels within the mitochondrial membrane, Mitochondrial Permeability Transition Pores (MPTP), open allowing proteins to move freely across the membrane [15]. The consequences of pore opening are osmotic stress on the outer membrane with eventual rupture; leading to the release of reactive oxygen species as well as release of mitochondrial proteins [13,16]. There is also further disruption of the mitochondrial function by the occurrence of intracellular Ca 2+ due to I/R injury where an excessive amount of Ca 2+ is accumulated in the mitochondria and this will result in uncoupling of oxidative phosphorylation and defect in ATP production [6]. Once again the loss of ATP perpetuates the altered ion gradient and degeneration of enzymes within cell [16]. It has been shown that the opening of the MPTPs in first few minutes of reperfusion is the critical determinant of the extent of I/R injury and can contribute up to 50% of the final size of infarcted myocardium [15]. Inhibitors of MPTPs therefore have been tested as potential therapeutic targets. The development of leaky mitochondria due to I/R injury generates the release of cytochrome C, proteases and caspases, which lead to apoptosis of the cell [6,16]. This process associated with the occurrence of intracellular Ca 2+ overload has been shown to contribute to the destruction of large amounts of cardiomyocytes upon re-establishing the blood flow in the ischemic myocardium [22], and is considered to be a major target for the prevention of I/R injury.
Depending upon the degree and duration of ischemia, the production of excessive quantities of Reactive Oxygen Species (ROS) takes place, lasting many hours after the restoration of blood flow [17]. Potent free radicals and oxidants such as superoxide anion, hydrogen peroxide, hydroxyl radicals, hypochlorous acid and nitric oxidederived peroxynitrite are generated. The postulated mechanisms for which the development of these oxyradicals and oxidants include the presence of xanthine oxidase post ischemia, activated neutrophils that migrate to the damage cardiomyocytes, electron outflow and cytochrome disruption from the leaking mitochondria. The release of a massive amount of catecholamines and their oxidation subsequently as well as the generation of cyclooxygenase and lipoxygenase contribute to the growing pool of ROS during I/R injury [3,6,17]. The ROS have many effects on the cardiomyocyte; they act at the molecular level by increasing Ca 2+ uptake in cardiomyocytes, influencing activities of sarcolemmal ATPases, activating of intracellular proteases and producing the breakdown of cellular structural components [3,6,17,18]. Superoxide and peroxynitrite have been shown to trigger DNA strand breakage, this subsequently activates nuclear enzyme Poly-ADP-Ribosyl-Synthetase-1 (PARP-1), a potent cause of infarction and contractile dysfunction in the heart [18]. The absence of PARP-1 in knockout mice showed less contractile dysfunction after ischemia [19]. ROS have also been shown to increase the activity of intracellular proteases such as matrix metalloproteinase-2 in the I/R hearts [20]. These proteases as well as the ROS themselves have destructive effects on phospholipids in membranes of the cell and structural components of the interstitial matrix [21,22]. The destruction of the sarcolemmal membrane by ROS is a major player in impairing the contractile function of the cardiomyocyte. There have been studies showing that scavengers of ROS such as superoxide dismutase, N-acetylcysteine and mercaptopropionylglycine help to preserve myocardial function [18,20]. Interestingly, some studies focused on ischemic preconditioning have also found that a decrease in the concentration of certain types of ROS is beneficial as it contributes to cardioprotection by decreasing the proteosome activity [13]. Finally, the formation of ROS has been shown to stimulate the inflammatory response through leukocyte activation, chemotaxis, and leukocyteendothelial adherence [23].
The generation of elevated intracellular Ca 2+ and ROS has been observed to activate intracellular proteases [24]. The activity of proteases contributes to I/R injury by altering the interplay between intracellular and extracellular proteins resulting in cardiac dysfunction [22]. The dysregulation of proteases interferes with homeostasis in the cell, generating misfolded or malfunctional proteins which affect myocytes by degradation of structural components, interstitial matrix, cell adhesion proteins, myofibrillar proteins and mitochondria membrane proteins [24]. Proteases also contribute to apoptosis in cardiomyocytes through the stimulation of signaling proteins directly as well as through the destructions of mitochondrial membrane. Cardiac proteases that have been shown to play a role in I/R injury are calpain, metalloproteinases, cathepsins as well as amino and dipeptidyl P peptidase-4. Several animal and human studies have shown a reduction in I/R-induced cardiac dysfunction when the inhibitors of the aforementioned proteases are present prior to and during the reperfusion phase of the ischemic heart [21].
Numerous studies have shown that I/R injury lead to altered expression of genes responsible for coding proteins that are important for the proper cardiac function. It has been shown that messenger RNAs (mRNAs) for proteins such as myosin heavy and light chain isoforms, myofibrillar Ca 2+ stimulated ATPase [25], Na+ -K+ ATPase isoforms [26] as well as mRNAs for sarcoplasmic reticulum proteins including ryanodine receptor, phospholamban and calsequestrin [27,28] were depressed due to I/R injury. It was proposed that depression of mRNAs due to I/R injury may be the consequence of oxidative stress as well as intracellular Ca 2+ overload in the I/R heart [25,28], which may induce these changes by affecting the transcriptional process in the nucleus.
The depression in mRNA activity due to I/R injury may also be caused by their modulation via micro RNAs (miRNAs), which represent a class of small endogenous noncoding RNAs that negatively regulate gene expression via degradation or inhibition of their target mRNAs. It has been proposed that miRNAs regulate more than 30% of the proteincoding part of the human genome [29]. Overall function of miRNAs is to alter cellular phenotypes via modulation of protein expression or to down-regulate protein expression when needed. Therefore, miRNAs are considered to be important regulators of cellular function in health and disease [29]. In the heart, miRNAs have been shown to play important roles in different physiological and pathological processes, such as cardiac development, myocyte contractility, cardiac fibrosis, arrhythmogenesis [30] as well as in the pathophysiology of myocardial infarction [31]. Emerging evidence indicates that ischemia induces profound changes in miRNA expression in different tissues/organs [32]. In myocardial ischemia, expression profiles of miRNAs may differ within different areas of the same ischemic heart, depending on the location of injury and the nature of the stimulus [32]. For example, it has been shown in the infarcted rat hearts that the expression of some miRNAs in the border area was much different from that in the infarcted area [31]. The roles of different miRNAs are very different in I/R injury; some of these such as miRNA-126, miRNA-133 and miRNA-144 have been shown to be cardioprotective whereas other miRNAs, including miRNA-1 and miRNA-15 have opposite effects. Furthermore, some of these like miRNA-21, miRNA-24 and miRNA-29 are proposed to be doubleedged in the processes underlying I/R injury [32,33]. It has also been reported that changes in expression of miRNAs occur in late stages of infarction (days after coronary occlusion), showing the possible involvement of microRNAs in cardiac remodeling and heart failure [34,35]. More recently, it has been documented that changes in miRNA expression occur also in the first few hours of I/R injury. Additionally, these changes may be either reversed or enhanced in hearts exposed to ischemic preconditioning as well as postconditioning prior to coronary occlusion [36]. Taken together, both mRNAs and miRNAs are important players in cardiac remodeling due to I/R injury and seem to be emerging targets of potential therapies in management of cardiac diseases.
Leukocyte recruitment, platelet aggregation and complement activation are also important factors that contribute to I/R injury after the blood flow has been re-established to the ischemic myocardium. The progression of lethal injury to the myocardium is partially mediated by the inflammatory processes, which contribute to the pool of destructive proteases and the generation of ROS. Activation of inflammatory mediators affects not only cardiomyocytes but also the vascular endothelium [37]. In experimental pre-clinical animal studies, the inhibition of neutrophil aggregation and activation has shown to limit the degree of I/R injury; however, the clinical trials have not proven to be as equally adventitious [38]. The degree of I/R injury is also correlated with the activation of platelets [39]. Complement activation in the reperfused heart recruits more inflammatory cells, compromises the endovascular function and impairs blood flow to the recovering myocardium [3]; this has been termed as the no-reflow phenomenon. From a more molecular point of view inflammatory mediators contribute to the injury of vascular endothelium through tumor necrosis factor-α and interleukin-1 during the development of I/R injury in the heart [40,41].
The clinical manifestations of I/R injury include myocyte hypercontracture, myocardial stunning, microvascular and endothelial dysfunction, arrhythmias and eventually cardiomyocyte necrosis and death. The degree of these abnormalities due to I/R injury lasts for different time intervals and their long-term impact is related mostly to the duration of ischemia before the induction of reperfusion [3]. The consequences of I/R-induced injury in the heart are described in Figure 1.
Myocardial hypercontracture is a direct result of the increase in intracellular Ca 2+ due to the impaired ion channels, specifically the Na+ -Ca2+ exchanger and the Na+ -H+ and Na+ -K+ ATPases in the sarcolemmal membrane [42]. Hypercontracture may be initiated by intracellular Ca 2+ overload as well as ATP depletion and contribute to myocardial injury by damaging cytoskeletal structures. The contracted cardiomyocytes tear away from adjacent cells breaking the intercellular junctions furthering the damage to the sarcolemma of the adjacent cells [43]. On the other hand, myocardial stunning is one of the main clinical manifestations of I/R; it is characterized by prolonged post-ischemic dysfunction of viable ventricle salvaged by reperfusion mostly due to the persistence of anaerobic metabolism [3,8]. Experiments have been carried out to show that myocardial stunning after 15 minutes of ischemia in healthy dog hearts can last up to 48 hours prior to the return of full contractility [43]. This has also been reported in humans after exercise stress test or balloon angioplasty where diastolic and systolic dysfunctions are seen from minutes to days afterward [44].
Figure 1: Schematic design describing the consequences of I/R injury in the heart.
The generation of ROS, activation of proteases and inflammatory factors are all contributors to microvascular defects and endothelial cell dysfunction in the no-reflow phenomenon. In fact, the recovery of the myocardium, specifically contractile reserve and function are dependent on the integrity and preservation of the vascular endothelium [45]. Endothelial cell dysfunction is characterized by impairment of vasodilatation and an exaggerated response to vasoconstrictors such as endothelin-1 and ROS, which reduce blood flow throughout the coronaries prolonging the ischemic time [46]. Additionally, endothelial cell dysfunction allows the expression of important mediators of the inflammatory response for neutrophils and platelets [47]. Platelets that become activated due to endothelial cell dysfunction contribute greatly to the size of infarct. When P-selectin, a cell adhesion molecule for platelets, knockout mice were compared with wild-type mice after certain periods of ischemia it was found that the mutants had significantly smaller infarct size then wild-type and that the number of activated platelets were directly correlated to the ischemic time [48]. Currently, one of the main stays of Myocardial Infarction (MI) treatment is aggressive antiplatelet therapies with aspirin, clopidogrel and glycoprotein IIb/IIIa inhibition.
Arrhythmias, particularly accelerated idioventricular rhythms, are common in patients during periods of post ischemia [49-51]. Arrhythmias usually occur during the first minutes of reperfusion but are rarely serious. Sustained Ventricular Tachycardia (VT) and Ventricular Fibrillation (VF) are usually secondary to prolonged occlusion as seen in the no-reflow areas [52,53]. The majority of the arrhythmias are caused by microvascular damage, stunning and swelling of cells and electrolyte imbalances within the cytosol of myocytes [54]. In particular, calcium influx into the cell post reperfusion and eventual restoration of ATP stores can cause the calcium dependent arrhythmias [55]. Angiotensin II (Ang II) receptors have also been implicated in the reperfusion arrhythmias; Harada et al. [56] showed that Ang II type 1a receptor (AT1a) knockout mice had less post ischemic arrhythmias compared to wild-type mice with similar infarct sizes. The wild-type mice also responded to AT1 receptor antagonist, which blocked the reperfusion-induced arrhythmias in the ischemic heart [56].
The last and most devastating effect of the reperfusion injury is cardiomyocyte death due to both necrosis and apoptosis. Some animal studies have revealed that up to 50 percent of infarct size can be contributed to the I/R injury [3,8]. This is due mostly to the destruction of intracellular organelles the swelling and rupture of cell membrane leading to necrosis. However, the degradation and release of mitochondrial proteins leads to apoptosis, a process that is more controlled and involves protein breakdown and DNA fragmentation [57]. The process of apoptosis in I/R hearts is a target for some therapies.
Our knowledge about the pathophysiology of I/R injury has significantly improved in the past 40 years but we have not bridged that with the ability to effectively reduce the I/R-induced complications in patients after MI. Currently, the most effective therapy is decreasing the total duration of ischemic time in patient with MI when subjected to coronary artery angioplasty or cardiac bypass surgery. However, some of the reasons that other interventions are less successful because of the multiple mechanisms involved and thus targeting anyone pathway during the development of I/R injury is an over simplification. Additionally, different therapies may have underwhelming results because their administration is not at the optimal time to have an impact on cardiomyocytes. Many of the pre-clinical studies that focus on animal models may not be accurately simulating those required for humans. Animals are also not administered the risk reduction medications, such as angiotensin converting enzyme inhibitors, beta-adrenoreceptor blockers or antiplatelet agents, prior and post induced ischemic events. These I/Rinduced events in animals are also performed under anesthesia, which may have some impact on the inflammatory response. In humans, patients have usually been experiencing angina prior to the injurious event, potentiating up-regulation of protective mechanisms such as nitric oxide or heat shock proteins. We also know that although the inflammatory cells that contribute to I/R injury are involved in the healing process, therefore targeting the inflammatory mediators and cells may have unwanted results. Nevertheless, there has been ample amount of research into several therapies to reduce or prevent the impact of I/R injury on the cardiac myocytes. Different current and potential therapies for the salvage of I/R injury are shown in Figure 2.
A main stay of treatment at the time of MI is to inhibit platelet activation. Activated platelets contribute to microvascular injury and continued ischemia [58]. These medications are also beneficial at the time of reperfusion as a means to minimize endothelial cell injury and reduce the no-reflow phenomena [59]. Another form of platelet inhibition is through glycoprotein IIb/IIIa inhibitors such as abciximab, eptifibatide, or tirofiban. A meta-analysis looking at glycoprotein IIb/IIIa inhibitors has shown that the addition of these drugs has improved ST segment resolution. However, animal trials using trapidil, a potent antiplatelet drug, are designed to impairing not only the activity of the platelets but also their migration to the injury area [60]. Liu et al. showed that rabbits who were exposed to different periods of ischemia and given trapidil, had a significant decrease in the ROSmalondialdehyde levels and increase in protective superoxide dismutase, as well as decrease in the expression of apoptotic signaling protein bax [61]. Antiplatelet drugs continue to be important in management of MI but no drug individually in clinical trials has shown large benefits in I/R [62].
There has been extensive research as well as clinical trials in the past thirty years investigating the effects of vasodilatory agents including adenosine on I/R injury. Adenosine has properties that make it an attractive therapeutic agent in this condition because it also serves as a substrate for the formation of ATP to replenish its diminishing stores in the ischemic myocardium. Adenosine is known to exert microvascular dilatation and reduce inflammation; the information gained from animal models has resulted in clinical trials [63-65]. Acute-Myocardial Infarct Study of Adenosine (AMISTAD) trial [66] has shown that treatment of the acute MI patients with adenosine was associated with significant reduction in infarct size, but only in anterior locations. In fact, the adenosine-treated group had a trend toward more major adverse events, such as; death, re-infarction, stroke and heart failure, as compared with the placebo group. Another trial that investigated the left-ventricular systolic and diastolic functions before discharge, showed no difference in placebo versus adenosine groups [67]. However, there was a trend toward lower mortality in the adenosine-treated group at 12 months. It is pointed out that the AMISTAD II trial investigated intravenous adenosine administration at low and high doses during primary reperfusion, fibrinolytic therapy or angioplasty, for acute anterior ST elevation MI versus placebo [68]. The nuclear imaging assessment of infarct size showed significantly lower areas in the high-dose group than placebo. However, there was no reduction in the combined clinical end point of heart failure or overall mortality at 6 months follow-up, but there was a trend toward fewer occurrences. The intracoronary administration of adenosine to patients undergoing angioplasty for acute MI was also investigated by Marzilli et al. [69] who found that intracoronary adenosine administration resulted in significantly lower creatine kinase levels and greater improvement in regional contractile function on echocardiography 1 week after the treatment. On the other hand, the PREVENT-ICARUS trial showed no benefit of pre-procedural intracoronary adenosine provided in terms of procedural myonecrosis [70]. Thus the evidence behind administration of adenosine either intravenous or intracoronary is varied and it appears that there is not enough support to use this agent as a mean to reduce clinical events or I/R injury at the time of PCI. On the other hand, some vasodilators such as sydnonimine, a class of Nitric Oxide (NO) donors, as well as Angiotensin Converting Enzyme (ACE) inhibitors have been shown to decrease the infarct size in animal models [71-73]. NO reduces infract size but whether this translates to an improvement in long-term functional benefit remains to be investigated. Nonetheless, ACE inhibitors have been shown to exert beneficial action post ischemic event, specifically scavenging free radicals and vasodilatation of the coronary arteries [74].
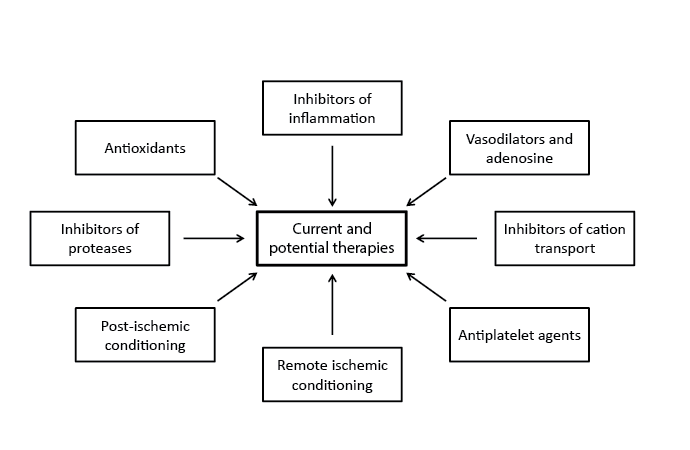
Figure 2: Outline of current and potential therapies in the management of patients with I/R injury.
Dysfunction of transmembrane ion channels is considered to be a major contributor to I/R injury mainly through changes in pH and intracellular Ca 2+ concentration. Therefore, these channels are potential targets for therapy to reduce the impact of reperfusion on cardiomyocytes. Na+ -H+ exchange inhibitors are helpful in the preservation of intracellular pH and Ca 2+ concentration. It was found that the inhibition of Na+ -H+ exchange in animals led to a reduction in infarct size [75]. A clinical trial with 100 patients undergoing PCI for acute anterior MI were administered cariporide, a selective Na+ -H+ exchange inhibitor, or placebo showed significantly better ejection fraction in the treated group versus placebo at 3 weeks and there was less myocardial enzyme release in treated patients [76]. However, another clinical trial including 1,389 acute MI patients undergoing reperfusion therapy evaluated the effect of eniporide, another selective Na+ -H+ exchange inhibitor, showed no improvement in clinical outcome for treatment versus placebo groups [77]. In a subgroup analysis there was a significant reduction in severe heart failure in patients who were reperfused more than 4 hours after onset of symptoms and received high-dose eniporide. Ranolazine, an inhibitor of late Na+ channels, has been shown to decrease Na+ dependent intracellular Ca 2+ overload in I/R injury [78,79]. This reduction in intracellular Ca 2+ has shown to be effective in chronic angina as well as decreasing the infarct size in acute MI [80]. A study by using a porcine model of I/R injury showed that ranolazine administered with or without propranolol improved left ventricular end-diastolic pressure and led to lower ROS production as well as more preservation of mitochondrial structural components including decreased opening of MPTP [81]. It should be mentioned that K+ ATP channels have been shown to improve outcomes in the ischemic preconditioning model where up-regulation of these channels aids in restoring the ion gradient and reducing the infarct size after true ischemic events [82]. Nicorandil, a K+ ATP channel opener, was investigated in a couple of small clinical trials, showing that nicorandil treatment improved regional wall motion abnormalities and was associated with fewer serious in-hospital events including arrhythmias and severe heart failure [83,84]. However, there needs to be more clinical trials investigating these cation pump inhibitors to better understand their effects in patients with acute MI.
Inhibition of the inflammatory process, specifically the activation and accumulation of neutrophils has had mixed evidence for reduction of I/R injury post MI [85,86]. This may be secondary to decreased drug availability to injury site or that the activation of inflammatory mediators has already occurred prior to the intervention. The complexity of the inflammatory process may also contribute to underwhelming improvements in function outcome in patients given treatment with these inhibitors. Inhibition of complement activation has also been investigated; two early clinical trials [87,88] showed no reduction of infarct size when patient were administered an anti-C5 monoclonal antibody, pexelizumab, as compared with placebo. More recently a clinical trial with 2885 patients at reperfusion post MI, were divided to receive either pexelizumab bolus or infusion versus placebo. There were no differences in a 30-day mortality observed in placebo versus treatment groups and also the incidence of shock and heart failure was similar in both groups [89]. This could be a result of late administration of the drug or lack of activity at the site of interest but either way further trail are unlikely unless a new method of delivery is developed. Another immunosuppressive agent that has shown to inhibit the opening of MPTP is cyclosporine; the effect of cyclosporine on MPTP is not selective and can also inhibit phosphatase calcineurin that may have larger more detrimental effects [90,91]. A small clinical study showed an intravenous dose of cyclosporine given prior to PCI decrease creatinine kinase and translated to a 20% reduction of infarct size in a subset of patient [92].
The role of ROS in the development of I/R injury has encouraged interest in the use of antioxidants as a means to scavenge ROS to attenuate their effects. Even though animal studies have shown to be promising, the evidence that antioxidant show clinical improvement in infarct size or decrease in heart failure is mixed [92]. Some of antioxidants that have been investigated are erythropoietin (Epo), estrogen, heme oxygenase and hypoxia induced factor-1 [93-96]. Epo has been extensively investigated, including a met-analysis in 2012 in which authors found 13 clinical trials with 1564 patients [96]. The authors found that administration of Epo did not improve left ventricular ejection fraction and that there was no effect on infarct size (as assessed by cardiac MRI). Epo group did not show a decrease in risk of adverse events or risk of heart failure, and all causes mortality was similar in both groups. The lack of clinical efficacy for antioxidants in I/R injury is most likely a result of the multiple mechanisms that contribute to this injury and attenuation of one pathway may not be clinically significant. Another reason may be that concentration of antioxidants at the infarction site may be decreased by microvascular injury when administered intravenously. In view of the rapid nature of ROS action for the induction of cardiac damage, the antioxidant therapy may not exert beneficial effects from the treatment viewpoint but instead may be of great value from the preventive viewpoint.
Most pre-clinical trials have focused on calpain and Matrix Metalloproteinases (MMPs), specifically by administration of their inhibitors prior to the induction of I/R injury. Calpain inhibitors such as MDL-28170 and SNJ-1945 have been administered in the animal models and have shown modest decreases in infarct size or improvement in the left ventricular function [97,98]. Common MMP inhibitors are doxycycline, 1-, 10-phenanthroline and GM6001. In a review by Hughes and Schulz [99] for the pre-clinical studies of MMP inhibition, a protective effect of inhibition of MMP-2 on left ventricular function was indicated. There was, however, a modest improvement in other clinical features such as myocardial performance, infarct size and mortality. MMP-2 inhibitors have been investigated in 4 clinical trials with the inhibitor doxycycline. None of them has shown any benefits in regards to left ventricular function or decreased infarct size [99]. A criticism of proteases inhibitors trials may be that there is a disconnection between pre-clinical and clinical investigations; specifically the pre-clinical trials address to replicate a clinical environment. For example most trials on animals perform a short to medium term MI which may not truly represent the I/R injury during thrombolysis or PCI. On the other hand, in isolated cardiomyocytes, inhibition of MMP-2 with siRNA has shown to be protective against I/R injury suggesting an important role of MMP-2 in contractile dysfunction in cardiomyocytes subjected to I/R injury [100].
In addition to different pharmacological therapies used in management of patients with I/R injury, it should be mentioned that the heart has its own potential to increase the tolerance to ischemia and reduce the severity of I/R injury by the activation of endogenous cardioprotective mechanisms. This type of protection was first demonstrated in 1986 by Murry et al. [101], who found that brief episodes of ischemia and reperfusion could protect the myocardium from subsequent prolonged ischemia, termed ischemic preconditioning. Since then, ischemic preconditioning-induced cardioprotection was observed in different animal species, including humans [102-104]. A broad scale of molecular mechanisms has been proposed to be involved in ischemic preconditioning including stimulation of adrenergic receptors [105], modulation of the activity of different protein kinases [106,107], release of NO [108] and production of ROS [109]. Additionally, it has been shown that the application of brief repetitive episodes of I/R reduces the extent of I/R injury also when applied during early reperfusion, termed ischemic postconditioning [110,111]. Different animal studies have shown that postconditioning has similar effects as preconditioning, involving the activation of survival protein kinases [112,113], whereas the protective effect of postconditioning includes decreases in total number of necrotic and apoptotic cells leading to smaller infarct sizes [114]. However, translation of ischemic preconditioning and post-conditioning to clinical settings is limited due to time limitation and invasiveness.
Another form of ischemic conditioning, remote ischemic conditioning, a term introduced by Przyklenk et al. in 1993 [115], when cardioprotection is achieved by ischemic insult on remote organ or occlusion of remote vessel in the same organ, seems to be the more promising strategy than classical ischemic conditioning. Further research revealed that the remote ischemic conditioning protection can be induced by ischemia of broad scale of distant tissues (either cardiac or non-cardiac) and evokes systemic protection against the acute I/R injury [116-118]. A growing number of experimental studies using animal models have demonstrated beneficial effects of brief limb ischemia in remote conditioning of the heart [119,120]. Moreover, this inter-organ protection was achieved by limb ischemia in humans [121]. Unfortunately, despite of the successful results achieved by different types of ischemic conditioning in experimental studies, the translation of these to clinical practice is limited and results are not conclusive. The most probable reason is that ischemic conditioning is a “healthy heart phenomenon” and failed in the case of patients with different comorbidities such as hypertension, hyperlipidemia, diabetes, insulin resistance, heart failure, altered coronary circulation and aging, as well as co-medications including statins, nitrates and antidiabetic drugs, that all together may modify the patient’s response to cardioprotective interventions [122].
I/R injury of the heart represent one of the leading causes of morbidity and mortality worldwide. Various procedures such as coronary angioplasty, cardiac by-pass surgery, cardiac transplantation, and thrombolytic therapy in addition to delayed opening of collaterals in the heart after an ischemic insult are associated with development of the I/R injury. This injury leads to impaired heart function and myocardial cell damage. Numerous mechanisms including occurrence of oxidative stress, Ca 2+ -overload, alterations in gene expression, increased activities of proteolytic enzymes and activation of inflammatory processes have been identified to be involved in the I/R injury and proposed as a potential targets for its therapy. Different pharmacologic agents such as anti inflamatory drugs, vasodilators, cation channel inhibitors and antioxidants have been tested in animal models as well as in clinical trials for the prevention and treatment of the I/R injury. Many of these agents have been shown more or less efficient in preventing the development of the I/R injury; however, their efficiency in the treatment of I/R is not conclusive. Therefore, development of the new pharmacologic agents with therapeutic potential in management of patients with I/R injury is needed. Although ischemic conditioning including pre- and postconditioning as well as remote conditioning seem to be the promising strategies for decreasing the negative consequences of I/R injury, their translation into the clinical practice did not produce satisfactory results to date. Nonetheless, the remote ischemic conditioning has been proposed to have emerging significance with respect of clinical interventions. Thus, revealing of molecular mechanisms included in remote ischemic conditioning may produce satisfactory results in translation of this endogenous cardioprotection to clinical practice in the near future. It is mentioned that Ibanez et al. [123] have also indicated a great success in reducing ischemic injury but have emphasized that the time has come to focus our efforts on future therapies for reducing the reperfusion injury.
The infrastructure support for this project was provided by the St. Boniface Hospital Research Foundation. Dr. Monika Bartekova was a visiting scientist from the Institute for Heart Research, Slovak Academy of Sciences, Bratislava, Slovakia.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation: Ducas A, Bartekova M, Dhalla NS (2015) Ischemia-Reperfusion Injury of the Heart: Moving forward with our Knowledge. J Hear Health 1 (3): doi http://dx.doi.org/10.16966/2379- 769X.110
Copyright:© 2015 Ducas A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access