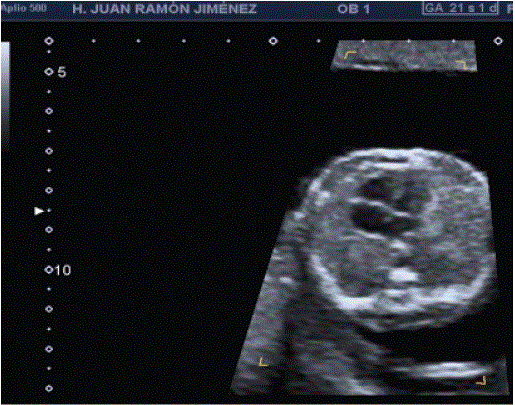
Figure 1: Shows an axial view through the area of cystic adenomatoid malformation of the left fetal lung, type II. Large left-sided lesion displaces the heart to the right.

Elga López González* Ana Sillero Castillo
Servicio de Obstetricia y Ginecología del Hospital Juan Ramón Jiménez de Huelva, Spain*Corresponding author: Elga López González, Servicio de Obstetricia y Ginecología del Hospital Juan Ramón Jiménez de Huelva, Spain, E-mail: elga_t@hotmail.com
The Cystic Adenomatous Malformation (CAM) is a rare fetal lung lesion. Its frequency is about 1 in 4000 births and is characterized by an overgrowth of terminal respiratory bronchioles that forms cysts of various sizes. It can be associated with polyhydramnios and pulmonary hypoplasia. We describe here a case of Adenoid Cystic Pulmonary Malformation type II with prenatal diagnosis at 20 weeks of gestational age, as well as its subsequent management. Morphological ultrasound describes multiple small cysts associated with a slight mediastinal deviation. The prognosis depends mainly on the associated injuries. Prenatal ultrasound allows early diagnosis and follow-up of CCAM to monitor the appearance of possible pathophysiological complications that may worsen its prognosis.
Prenatal diagnosis; Lung; Cystic adenoma
Pulmonary Cystic Adenoid Malformation (CCAM) is a rare anomaly in lung development with few cases reported in the world literature. It was first described by Bartholinuis in 1687, but was not described in more detail until 1949 by Chin and Tang. Cystic adenomatoid malformation of the lung (CCAM) is an adenomatous proliferation of the terminal bronchioles, with suppression of alveolar growth [1,2].
It is an uncommon malformation. The clinical presentation is highly variable. The incidence ranges between 1/11,000 and 1/35,000 of newborns [3]. In most diagnosed cases the lesion is unilateral (generally in the left lung and in a single lung lobe) [4]. Together with the bronchopulmonary sequestration and bronchogenic cyst, it is part of a group of anomalies based on dysplasia in the embryonic formation of the pulmonary tree. The diagnosis of CCAM is ultrasound [5] and is usually made at early gestational ages.
To know in depth that the adenoid cystic pulmonary malformation, focusing the search on the classification of the entity and its associated prognosis with a view to good prenatal advice.
A bibliographic search was carried out through PubMed, The fetus. net, Scielo.
Secondary pregnancy without data of interest derived in the first pregnancy visit (11+6) due to megabladder without observing other alterations. It has normal translucency and low-risk screening for chromosomal diseases. At 16s, persistence of the megabladder (diameter 16 mm) and mild bilateral pyelocalyceal dilation were observed.
In the Morphological ultrasound of the 20th week, no alterations in the urinary system were observed, but a slight dextroposition and levo-mesoapex were observed in the thoracic examination (Figure 1). In the left hemithorax, echogenic formation with some cystic area, approximately 18 × 15 × 23 mm (Figure 2), avascular, without being able to demonstrate the nutrient vessel that moves the heart to the right. No signs of hydrops are observed.
Figure 1: Shows an axial view through the area of cystic adenomatoid malformation of the left fetal lung, type II. Large left-sided lesion displaces the heart to the right.
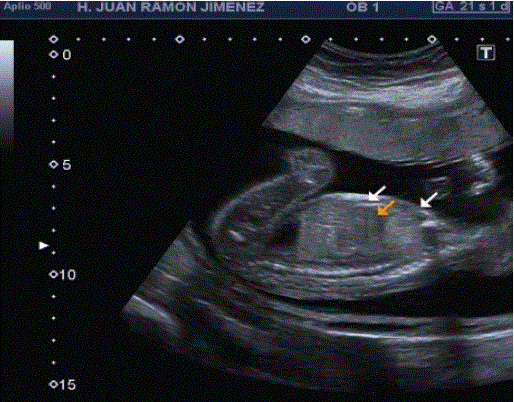
Figure 2: Sagittal views of the cystic adenomatoid lesion of the fetal lung (arrow).
A cytogenetic study is performed using a conventional karyotype in amniotic fluid and microarrays; with normal result of both.
At 24s, a consistent biometry is noticed in the thoracic examination, an echogenic formation is observed at the left lung level of 28 × 24 × 20 mm, with cystic areas inside (Figure 3). In this lung, a normalappearing marginal posterior reed is evident. Mediastinal deviation (dextroposition, mesoappex) (Figure 4). There is no evidence of hydrops fetalis or polyhydramnios.
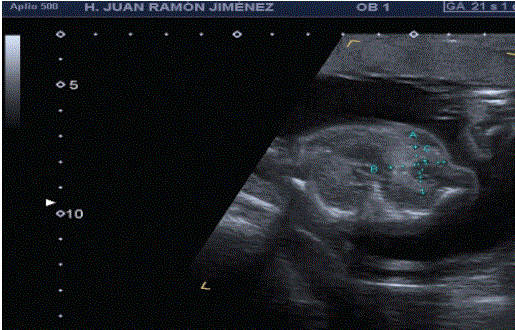
Figure 3: Cross section of the thorax where we can see hyperechoic formation at the level of the left lung with cystic areas inside.
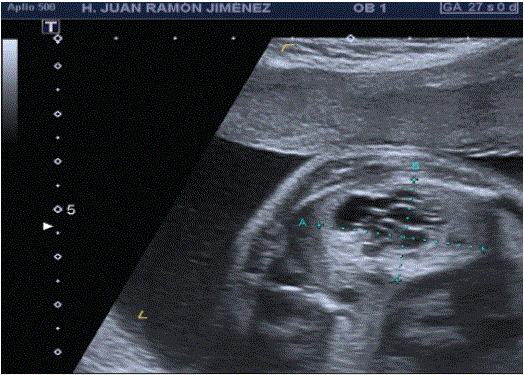
Figure 4: Cross section of the heart: dextroposition and mesoápex.
The ultrasound controls were followed in the 27, 29, 32 and 35 where the patient reported not noticing fetal movements and perception of occasional contractions. The ultrasound describes a single fetus in the head with absent heart and body activity. Placenta: left lateral fundus. Amniotic fluid: decreased. ILA 5 PFE1724
The fetal autopsy describes a type II cystic adenoid malformation, with multiple cystic areas in the lower lobe of the left lung; heart in dextroposition; normoposition of vessels and megabladder. No hydrops fetalis or other alterations.
In the placental examination, intra and intervillous fibrinoid deposits stand out, with foci of vascular thrombosis and data compatible with chorioamniotis.
The first description of the CCAM was made by Chin and Tang in 1949. The microscopic criteria were later defined by Kwittken and Reiner in 1962. Stocker et al made in 1977, on a review of 38 cases, the most widely used classification of this malformation, which distinguishes three types [6].
Large, single or multiple cyst (greater than 2 cm, reaching 4 to 10 cm, constitutes 50 to 65% of cases, frequently producing mediastinal displacement, and it was considered a good prognosis.
Multiple small cysts (less than 1 cm). The prognosis depends on the existence of associated injuries.
Voluminous and solid lesion that produces mediastinal displacement, formed by multiple millimeter cysts (<0.5 cm). Its association with other malformations is rare (10%). It is also associated with a poor prognosis.
CCAM is the result of a blockage in cell development in an early stage of the embryo [7,8]. The proliferation of glandular tissue without alveolar differentiation produces adenomatoid histology. The arterial and venous connections are typically normal, but the bronchial system is not defined [9]. The type III CCAMs probably occurs between six and eight weeks after conception, since at six weeks the endodermal tissue derived from the terminal bronchioles induces in the tissue splanchnic mesenchymal the beginning of the development of the components of the bronchopulmonary segments. The pseudoglandular period (5 to 17 weeks) is so named because, histologically, lung development has a gland-like appearance. This is the period of development of the bronchiolar division with the differentiation of the air conduction system. The canalicular period (16 to 25 weeks) is reminiscent of type II CCAM. During this period, the lumen of the bronchi and bronchioles becomes larger and the lung tissue begins to be well vascularized [10].
It has been suggested that CCAM is due to a defect in lung maturation, perhaps due to failure or asynchrony in the interaction of the mesenchyme with the epithelium [11]. Embryologically, there would be a failure in the connection between the airway and the respiratory component, giving as Excessive growth of the terminal bronchioles resulted, without alveolar development. Local alterations in the production of certain growth factors that modulate normal lung development.
It has been suggested that CCAM is due to a defect in lung maturation, perhaps due to failure or asynchrony in the interaction of the mesenchyme with the epithelium [11].
Embryologically, there would be a failure in the connection between the airway and the respiratory component, resulting in excessive growth of the terminal bronchioles, without alveolar development. Local alterations in the production of certain growth factors that modulate normal lung development could be responsible for the altered lung structure observed in these lesions [12-14].
The natural history of this injury is variable. In general, the factors that are associated with a worse prognosis, and that can be identified by prenatal ultrasound, are cardiac and mediastinal displacement, polyhydramnios, ascites, and especially non-immune hydrops fetalis.
Prenatal reduction in size and even the disappearance of the lesions before delivery have been described [1,15,16]. This fact has been attributed to a combination of several factors, such as the actual decrease in size, the growth of the surrounding normal lung without change in mass size or loss of fluid/tissue interfaces, thereby reducing echogenicity. Neonatal survival is related to the presence or absence of pulmonary hypoplasia (conditioned by compression of the normal lung by the expanding mass) and associated abnormalities.
Periodic surveillance may be a reasonable option in those cases in which a reduction or absence of growth of the mass is observed [17], and provided that the aforementioned evolutionary complications do not appear. In cases of progressive lesion, especially if hydrops fetalis appears, intrauterine therapy (thoracentesis [18], thoracoamniotic shunt [19] and fetal laser surgery) comes into consideration, the end of gestation will be established according to the gestational weeks and complications.
Criteria for fetal treatment. The criteria to indicate treatment are: a) marked mediastinal displacement and b) frank signs of venous return disorder (Doppler of the ductus venosus with reverse atrial flow and/or tricuspid regurgitation). The third criterion is obviously the development of hydrops signs, but the ideal would be to act early before your debut, although this is not always possible. There is no good prognostic predictor for large masses, and although it has been proposed to use a ratio between the volume of the mass and the fetal circumference [20], most clinical groups usually rely on strict followup, generally weekly.
The objective is to avoid fetal death and to reach neonatal surgery without hydrops, which has a good prognosis. The technique of choice whenever there are cysts is the placement of a shunten in the largest cavity or area with greater cavities. There are no published series but there are various clinical cases in clinical series or as isolated cases [19,21,22]. In the author’s experience, in 5 treated cases, all with hydrops, the shunt allowed hydrops reversal and prenatal survival in all of them. Very exceptionally, in solid masses with a nutrient artery, interstitial laser coagulation has been described.
Finally, there are some recent studies that suggest that maternal corticosteroids could be associated with a reduction in the mass, and cases of resolution of the hydrops have even been described [23,24]. Although the results must be confirmed with larger series-there are 9 published cases-given the low aggressiveness of the treatment, it would seem reasonable to try treatment with corticosteroids and wait a few days for possible improvement.
The long-term evolution of CCAM and pulmonary sequestration suggest that most cases have good postnatal lung function [25,26]. Children who survive unshunt placement have good lung function and a slight increase in mild deformation abnormalities of the chest wall [27].
The clinical case described, corresponding to type II CCAM, the pathological anatomy describes a multicystic lesion located in the left lung, and signs of respiratory distress at necropsy with no other relevant data, except for hypoplasic morphology of the rest of the left lung and generalized visceral immaturity. In this case, the cause of intrauterine fetal death is the chest compression caused by the injury, without having a typical evolution of a complicated case (10% of cases) with the absence of hydrops and polyhydramnios, therefore without systemic repercussions.
Prenatal ultrasound allows early diagnosis and follow-up of CCAM. In the first place, other fetal malformations, which markedly darken the prognosis, must be excluded, obtaining a fetal karyotype if possible. After excluding other diagnostic possibilities, the evolution of the size of the mass and the appearance of possible evolutionary pathophysiological complications (mediastinal displacement, hypoplasia of normal lung tissue, polyhydramnios, cardiovascular compromise, hydrops fetalis) should be especially monitored. In the absence of complications, ultrasound surveillance is recommended, and let the gestation evolve until term. After delivery, a careful neonatal evaluation should be performed. The presence of respiratory distress will require immediate intervention.
Download Provisional PDF Here
Aritcle Type: CASE REPORT
Citation: González EL, Castillo AS (2021) Pulmonary Cystic Adenomatoid Malformation, Prenatal Diagnosis and Case Management. Gynecol Women’s Health Res 3(1): dx.doi.org/10.16966/2689-3096.119
Copyright: © 2021 González EL, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access