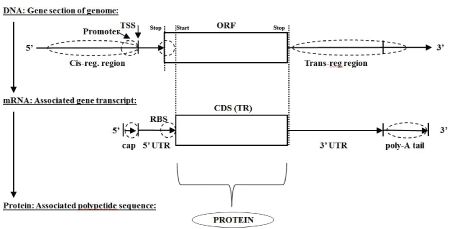
Figure 1: Classic Central Dogma: DNA mRNA Protein (monicistronic).

Stephen Winters-Hilt*
Computer Science Department, Connecticut College, New London, USA*Corresponding author: Stephen Winters-Hilt, Connecticut College, Computer Science Department, 270 Mohegan Ave, New London, CT 06320, USA, E-mail: swinters@conncoll.edu
Analysis of eukaryotic transcriptomes is done using BLAST validated mRNAs from Genbank. Each mRNA transcript is traversed with a simple ORF finder with three frame passes on forward reads and three frame passes on reverse compliment reads. In doing so we arrive at an encoding overlap-topology analysis of eukaryotic nucleic acid (transcriptome) sequences that parallels a previous analysis of prokaryotic nucleic acid (dsDNA genome) sequence (summarized in the Supplement). A reverse frame pass for the prokaryotic genome was necessary because the sequence information is only the reference ssDNA strand, requiring a second three-way frame pass for the reverse compliment ssDNA that completes the actual dsDNA prokaryotic genome. When the reverse frame pass is also done for the eukaryotic transcripts there is seen an overlap encoding topology like that seen in the intron-less prokaryotic genome. Even if the antisense overlap encoding in the eukaryotic transcripts is entirely non-functional, it indicates an intron-less archaeon/prokaryotic evolutionary artifact consistent with the viral eukaryogenesis hypothesis (summarized in the Supplement). In the Discussion, some of the eukaryotic reverse complement transcript encodings are thought to be functional given their lengthy miRNA signaling regions, suggesting a possible non-RNAi role for RNA-dependent RNA polymerase in eukaryotes.
Transcriptome Analysis; RdRp; Informatics; Viral Eukaryogenesis; RNAi; Intron Earliest
Healthy eukaryotic cells are known to have RNA-dependent RNA polymerases (RdRp’s) [1-8]. The native role of specialized RdRp’s in production of siRNAs is already widely understood [1-4], especially in the analysis of plant transcriptomes [1,3,4]. Plants are found to regularly use RdRp to amplify siRNA for RNAi defense processes [4] (possibly an essential mechanism for transposon control). A sophisticated role for RdRp-control in post-transcriptional gene-silencing (as a ratelimiting factor) has also been demonstrated, such as with N. crassa and D. melanogaster [9-11]. The study of RdRp in eukaryotes has been complicated since the early 1980’s, however, since in many cases their source could simply be attributed to viral origins [12]. Part of the mystery of the eukaryotic source for RdRp in some organisms is found to be simply a matter of RdRp being induced from an existing DNA-dependent RNA polymerase (DdRp), as is found to exist in a growing number of organisms [1] (there are also situations where RdRp can act as DdRp via transcription factor control [2]). Pol II is an example of a DdRp that can shift to being an RdRp, as seen in plants. In humans Pol II RdRp activity allows for novel regulation mechanisms [13]. In yeast (S. cerevisiae) Pol II is even involved in gene loop topologies in the complex early stages of transcriptional activation, where the Pol II DdRp binds and juxtaposes promoter and terminator ends of transcription at activation-indicating both ends of the transcriptional unit must be properly recognized by entering a loopconfiguration with Pol II at initiation of transcription [14].
The role of RdRp may be critical to eukaryotes in a variety of ways, including robust development, where some plants require RdRp for healthy, competitive, growth [3]. Likewise, yeast (S. pombe, N. crassa, and S. cerevisiae) and lower eukaryotes (C. elegans, A. thaliana, and D. melanogaster) are well-known to have RdRp activity [9-11]. RdRp activity has also been reported in rat brain cells [15] and rabbit reticulate cells [16]. In a study of axolotl [8], an evolutionarily conserved enzyme activity with properties of RdRp, but not RdRp II or III, is observed. In bats there even appears to be an instance of ‘recent’ RdRp gene adoption from virus [5]. Even if the previous Bat RdRp was of non-viral origin, numerous instances are thought to exist where enzymes in eukaryotes are replaced by (better) viral counterparts [17].
Extensive RdRp activity has been documented in Trypnosoma brucei (the unicellular parasitic kinetoplastid that causes African sleeping sickness) [18,19]. In T. brucei there are found the negative strands of the mRNAs of a number of genes, including: cytochrome b, cytochrome oxidase I, cytochrome oxidase III, and MURF 2 [19], indicating significant RdRp activity resulting in antisense transcript production. This is an example where the antisense encoding overlaps with a transcript produced with a positive sense encoding already present. Further indication use of negative sense strands, e.g., reverse complement encodings with respect to a reference ‘positive’ transcript, is described in the Results. The implications of this for RdRp processing in eukaryotes, spliceosome processing, and viral origins (where both of the prior methods may have been introduced), are discussed.
The possible ancient viral origin of RNA polymerases in eukaryotes is described in the study by Iyer LM, et al. [6], where there is a hypothesized loss of RdRp in the ancient cell line that is regained from virus in later eukaryotic cell lines for cytosol RNAi support (via siRNA production). It’s possible that the introduction of RdRp processes into both the cytosol and the nucleus of the proto-eukaryotic cell line were via the viral endo symbiont that is hypothesized to form the proto-eukaryotic nucleus, e.g., RdRp’s adoption could be a remnant of the hypothesized viral eukaryogenesis event itself. The origin of many critical nucleic acid processing enzymes, the transfer of nucleic acid processing methods in particular, appears to have been dominated by viruses transferring their methods to cellular hosts and rarely the other way around [17]. Perhaps the spliceosome is viral in origin as well or at least co-evolved with an ancient archaeon/prokaryote host. If the latter, however, we would expect to see spliceosomal activity in archaeons/prokaryotes, but there is no evidence of such. So simpler is to hypothesize that the list of hypothesized nucleic acid processing methods of viral origin, like RdRp, mRNA capping, and mRNA polyadenylation, also includes spliceosomal machinery. Details in support of this include spliceosomal introns (e.g., not the self-splicing Group I and II introns) are not seen in prokaryotes or archaeons, but are seen in viruses large enough to bother with the complexity [20], where the mega viridae class of viruses includes viral genomes larger than some cellular genomes [20-22], and in Mollivirus (infects Acanthamoeba), for example, has about 4% of its genes with spliceosomal introns.
Viral-based biomolecular machinery for passing virus molecules, including the viral genome, into a target nucleus is well-studied as it is a fundamental trait of all eukaryotic viruses [23-29] the exception being those viruses, usually very small, that attack during mitosis when the nuclear membrane is temporarily disassembled (the parvo virus, for example, is only about 5,000 bases long) [23-29]. So, viral machinery for nuclear transport is well known and it could have been prevalent and co-opted in a mutualism context, where a host cell having multiple large viruses and multiple prokaryotic endo symbionts could have existed in a proto-eukaryote then as it is does in amoeba now. Viral control of access to the present-day eukaryotic nuclear envelope [23-29] or possibly the nuclear-like envelope of another virus in a large cellular host [20]; viral control of the spliceosome [30,31] and viral encoding of spliceosomal molecules [20,32] for optimized processing on viral encoded genes that require spliceosomal processing [20,32] all lay the groundwork for a possible viral origin for the spliceosome in eukaryotic cells.
The standard hypothesis for the origin of the spliceosome in non-viral eukaryogenesis hypotheses is that it evolved from self-splicing (Group I and II) introns imported from the prokaryotic endosymbionts. And such a non-viral origin for the spliceosome has been suggested in the viral eukaryogenesis context as well [33]. The Results presented here appear to favor a viral origin for the spliceosome via the viral eukaryogenesis hypothesis. Results are shown that indicate an operational spliceosome in the proto-nuclear envelope as non-nuclear genomic information was being ‘adopted’, e.g., the spliceosome was under control of the protonucleus during the proto-eukaryote’s uptake of the non-nuclear (archaeon and prokaryotic) genomic material. The spliceosome likely evolved from the Group I and II self-splicing introns, as suggested by others [33], but this spliceosome development may have occurred at a much earlier time than viral eukaryogenesis. This is hypothesized to be the case given the sizable percentage of the eukaryotic transcriptomes’ (5% to 15% of mRNA transcripts) that have overlapped encodings that represent coding artifacts that are ‘intron-less’ in origin, e.g., such as would occur with an archaeon/ prokaryote ancestor having dually encoded transcriptome material (imprinted from dually-encoded intron-less genome). Some of the overlap encoding artifacts even appear to be functional given their long ORF encodings (greater than 300 bases) and long 3’UTR regions (greater than 200 bases), indicating a non-RNAi use for RdRp. A transcript-level (intron-less) overlap encoding is obtained from an unspliced-intron precursor via spliceosome processing. Further details and subtleties of the spliceosome and its possible viral origin [34-55] are described in the supplemental section.
The viral eukaryogenesis theory leaves little opportunity for testing since it relates to a hypothesized historical event, and for this reason testing the viral eukaryogenesis theory has been grouped with other hypothesized endosymbiosis events, such as those endosymbiosis events leading to the mitochondria and chloroplasts [56]. The Viral eukaryogenesis hypothesis, however, involves a shift of core (non-eukaryotic cytosol) genomic data into the newly formed nucleus of the organism, so may leave more evolutionary artifacts in the genomic sequence information than could be obtained in the case of the organelles.
Antisense encoded mRNA information is known to exist [19], in the Results, however, we see anomalous amounts of overlapping antisense encoding, with overlapped transcriptome encoding at percentages seen in some prokaryotes and archaea. Even if the antisense encoding aren’t functional mRNAs in their own right, they are artifacts of such, and in sufficient numbers to indicate the non-eukaryotic (non-spliceosomal) genomic information was imported into the proto-eukaryotic nucleus by way of transcriptome adoption, apparently favoring uptake of a dominant transcript. If any of the antisense encodings are functional this would then require use of an RdRp. Such use of RdRp is already well known to exist, however, for shorter, partial transcripts, where it is used to get siRNA for RNAi regulation and control. Thus a role for RdRp that is not RNAi related is suggested in the Results, in order to access “ghost” antisense transcripts from the ancient prokaryotic/archaeal ancestor, and to allow for the overlap encoding in general.
To understand the nature of the imprint artifact from the prokaryotic/ archaeal genome/transcriptome to the eukaryotic transcriptomes, it helps to first review some background on genome and transcriptome coding topology (as mentioned in Section-Genome and Transcriptome Coding Topologies and Central Dogmas), and overall classic central dogma for archaeons/prokaryotes. Since archaeons/prokaryotes very rarely exhibit introns (some do have self-splicing introns, however, and also for some viruses too [42]) their transcriptome structure almost directly maps to their genomic structure.
The classic central dogma of biology describes how information encoding in the form of a DNA polymer (or collection of DNA polymers, e.g., chromosomes) transcribes to messenger RNA (mRNA) polymers, which are then translated to protein using a three-base encoding scheme (see figure 1 for monocistronic example, for polycistronic see Supplementary figures 1 and 2, and for a simple informatics detection of the three-base encoding scheme see Supplementary Section-ORF overlap topology in prokaryotic dsDNA and Supplementary figure 3). The three-base encoding scheme leads to the discovery of anomalously long ORF encodings (as mentioned in Supplementary Section-ORF overlap topology in prokaryotic dsDNA and Supplementary figure 4), which is the basis of the ORF-finder algorithm described in the Methods section (with modifications for use with mRNA or EST data).
Figure 1: Classic Central Dogma: DNA mRNA Protein (monicistronic).
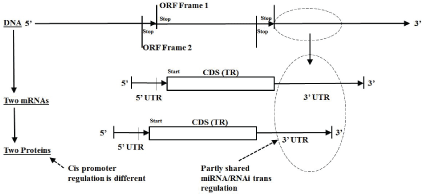
Figure 2: mRNA Transcriptome Topology Schematics: Overlapping monicistronic
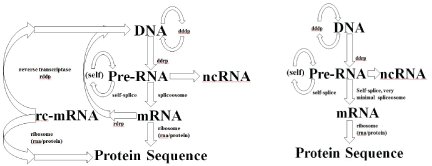
Figure 3: (Left) Modified Central Dogma (with reverse transcriptase and RdRp). Shows the modified central dogma with both reverse transcriptase and RdRp present in the evolutionary process, early cells are hypothesized to have lost their spliceosome and RdRp processes to arrive at the familiar prokaryotic cell, as shown in Right. (Right) Standard Central Dogma for Prokaryotic Cells. Minimal splicesome indicated, zero for many. Early cells eventually lose spliceosome and RdRp, but still have self-splicing introns to some extent.
For an organism’s genomic DNA information repository the transcription from DNA to mRNA can be done in multiple ways from the same section of DNA, e.g., different overlapping reads are possible where different three-base codon framings step across the same DNA encoding region (Figure 2 and Supplementary figure 5). For some prokaryotic organisms this overlap encoding can be quite significant (Supplementary figures 6 and 7, which relates to results given in a study by Winters-Hilt S (2006) [57]), and this overlap encoding by frame shift is effectively doubled for duplex DNA genomes (with encoding in the complement strand), as well as duplex RNA genomes, and ssDNA and ssRNA genomes that have an antisense (reverse complement) encoding read-out by way of an intermediate duplex form [42]. So, for prokaryotes a high degree of overlap encoding, for both forward and reverse reads, is already prevalent at the level of the genome (Supplementary figures 5-7), whether the genome is single stranded or duplex, or DNA-based or RNA-based.
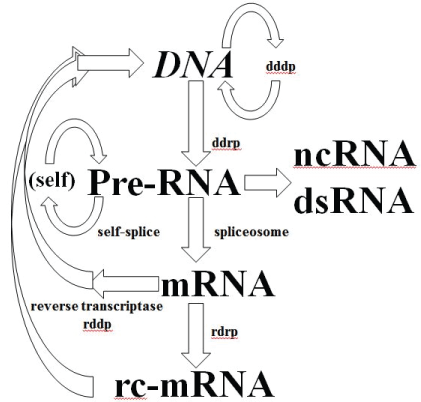
Figure 4: Central Dogma for Viruses (have RdRp).Virus has minimal cytosol, no ribosome, no protein production, since this is taken from cell, as is much of the DNA processing. Virus retains RdRp and reverse transcriptase (RdDp, DdDp), RdRp, (so nucleic acid polymerases), and spliceosome.
In Supplementary figure 7, reprinted with permission from the study by Winters-Hilt S (2006) [57], we see that the C. trachomatis genome is half coded on the forward strand and half on the reverse, with very little dual overlap encoding. In table 1 of study by Winters-Hilt S (2006) [57], for ORFs>200 bases in the V. cholera genome, the percentage with dual encoding is 6.57% (dual occurs when overlapping opposite read directions, with scores like 11000, 12000, and 21000 in Supplementary figures 6 and 7). For Deinococcus radiodurans (Supplementary figure 7) the amount of dual encoding is 69.4%.
| Species | #mRNAs | Download Date | Source |
| Mus musculus (Mouse) | 98484 | 8/19/2016 | NCBI genbank |
| ThunnusThynnus (Tuna) | 10163 | 7/5/2016 | NCBI genbank |
| Salmosalar (Atlantic Salmon) | 498523 | 7/7/2016 | NCBI genbank |
| Caenorhabditiselegans (Worm) | 8327 | 8/18/2016 | NCBI genbank |
Table 1: mRNA dataset download dates/versions
Once the prokaryotic transcription to mRNA is complete a selection for the RNA-based coding region is effectively done, and unique protein products are thereby directly indicated (see figures 3 and 4 for variations indicated on central dogma). There can be multiple protein products because prokaryotes can have polycistronic transcripts, whereby a single mRNA may have multiple, sequentially located (non-overlapping), regions that each encode their own protein product (a.k.a, operons, Supplementary figure 1). Eukaryotic transcript processing is more complex due to introns and alternative splicing (as mentioned in the Section-Alternative Splicing in Eukaryotes and Supplementary figures 8-10 for details). Once a eukaryote reaches the same stage of having a ‘mature’ mRNA, on the other hand, the resulting encoding is typically monocistronic (with cis-regulation governing only one encoding region). Simple eukaryotes, such as C. elegans, are known to have both types of encoding (monocistronic and polycistronic) in significant numbers [42].
The process of DNAmRNAProtein production is regulated at both transcription and translation polymerase stages. Cis regulation dominates at the DNAmRNA polymerase stage, and trans-regulation at the mRNAProtein polypeptide production stage. In the case of the polycistronic encodings, there is one cis-regulatory region for multiple coding regions (Supplementary figure 1). The dominance of transregulatory mechanisms at the mRNAprotein stage is significant because all living processes, including viral processes, can be regulated at this stage, and many of the regulatory processes involve simple antisense nucleic acid molecular recognition, indicating a possible common and ancient (RNA World) biomolecular process. In eukaryotes the process of DNAmRNAProtein production is also regulated at the spliceosome level (for which a brief background is given in the Section-RNAi). The main mechanism for trans-regulation in eukaryotes is RNAi. The role of trans-regulation in prokaryotes involves a non-RNAi process that employs no RdRp for siRNA amplification, using a method evidently separately evolved: CRISPR/cas [45,46,58,59].
Viruses are a well-known exception to the central dogma and have already modified the central dogma when it comes to the role of reverse transcriptase (Figure 4). Using reverse transcriptase, as the name suggests, it is possible to go back from RNA to DNA, altering the cell’s genomic repository of information (in an inheritable way even in multi cellular eukaryotes if that genomic DNA happens to be in a gamete cell). Viruses also have their own means for producing (or replicating) RNA information by way of the aforementioned RNA-dependent RNA polymerase (RdRp), while the production of pre-mRNA from DNA in the central dogma is by way of a DNA-dependent RNA polymerase (DdRp). The viral information processing, thus, includes everything in the central dogma up to the protein-production (cytosol) stage, as shown in figure 4. Further background on early cell/virus hypotheses that are consistent with the form of the viral eukaryogenesis hypothesis suggested here is given in the study by Winters-Hilt S [34].
Although RNAi has been discovered only recently [60,61], and is thought to have only become significant with the introduction of eukaryotes, some aspects of RNAi appear to be ancient and even fit within the RNA world paradigm. RNAi appears to be a universal process used by eukaryotes and their viruses, but not prokaryotes. A key component of the RNAi process is the use of a miRNA that is incorporated into a collection of argonaute proteins in what is known as the RISC complex [61]. The miRNA provides the nucleic acid sequence template that is used in regulating specific antisense-related mRNAs (Figure 5) [62]. The miRNA’s guide strand provides an antisense match to its mRNA target and plays a role in regulation or complete inhibition (if the target ssRNA is viral or a transposon). miRNAs may have had an early (RNA World) role as siRNAs given the discovery of a simple biogenesis pathway for miRNAs: ‘mirtrons’ are introns that once spliced anneal to themselves to form a miRNA. Mirtrons and mirtron-based RNA interference are also consistent with a “follow the introns” analysis (Supplementary file and [34]) that suggests that introns might have been fundamental and ancient. The appearance of RNAi throughout the eukaryotes is an indication of its ancient eukaryotic origin at least, if not even more ancient.
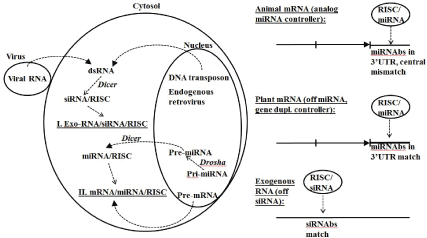
Figure 5: RNA interference (RNAi) defense and expression regulation in eukaryotes. Drosha clips pre-miRNA from mRNA form to the pre-miRNA form for export to Dicer. Dicer targets dsRNA intermediate (transposon, viral, and pre-miRNA) and creates miRNA or siRNA template and recruits RISC complex to hold ssRNA specificbinding template. When RISC complex with template binds RNA target have cleavage or binding according to specificity/stability of template match. A central miRNA template and miRNA binding site (miRNAs) mismatch leads to gene down regulation, while a perfect match results in a gene or ncRNA knockout. Strong binding site match region is 7-8 bases in length. Strong match, length of match, vs. mismatch. Epigenetic machinery of cell is partly regulated by a subgroup of miRNAs known as epi-miRNAs [63].
RNAi is an RNA interference method that requires formation of a dsRNA intermediate. In ssRNA gene-silencing experiments in 1998 [60], it was found that dsRNA was not as effective as ssRNA at silencing, and that the ssRNA silencing was “too good” in that a small amount of ssRNA would accomplish complete silencing, indicating an enzymatically catalyzed silencing process. The RNAi process was clarified in 2001 [61] with the description of Dicer and its role in preparing 22bp dsRNA segments for ssRNA template loading into the RNA-induced silencing complex (RISC). RNAi interferes with specificity by using ssRNA strand that is antisense to the ssRNA target of interest (an mRNA or a retrotransposon RNA intermediate, for example). The current form of RNAi may not have been around with the early eukaryotes. Early eukaryotes may have had a protoRNAi that was only mirtron based, and may not have had a significant RISC complex developed yet [63].
In the viral eukaryogenesis hypothesis to be discussed in the SectionViral Eukaryogenesis Hypothesis, we see that an RNAi interference CRISPR/cas precursor is hypothesized to have existed in the archaeon/ prokaryote-like cell (possibly with mitochondria) when it is invaded by a membrane-bound viral endosymbiont, where the membrane-bound invasion of the virus involves an RNA interference arms race (whereby the viruses membrane can protect and complete critical mRNA processing prior to release into the prokaryote-like host’s cytosol). Thus, the virus endosymbiont may have resulted from a mutually beneficial symbiosis (mutualism) on the basis of RdRp usage and RNAi co-evolution, that eventually resulted in the wholesale adoption of the viral machinery and separate nucleus processing in the hypothesized viral eukaryogenesis (discussed further in the Section-Viral Eukaryogenesis Hypothesis).
The mRNA data used in the transcriptome analysis is from the NCBI Genbank entries with the download dates indicated. The files are downloaded from www.ncbi.nlm.gov, where the mRNA database is selected, with search on the indicated organism, and download as file option selected. For the tuna and salmon data, the entire collection of sequences available on that day were used in the analysis (Tuna only had 10163 transcripts on 7/5/2016.) See table 1 for dataset download dates/ versions. For the mouse and worm data, both involving test subsets of the full mRNA dataset available (from the most recent mRNA listings), where the number of sequences used in the analysis is as shown. For mouse, the actual full set of mRNAs number about 50 times more than that examined, and a more comprehensive analysis is to follow. For worm, the test chunk was also a small fraction of the mRNAs available, and was mainly done to see how the acquisition of 3’UTR regions is expected to fail as richer operon structure, with ORFs frequently shorter than 300 bases, begins to become prevalent (as in worm). The results for worm indicate a very simple fix via a second pass of processing (but this is part of a separate analysis so not discussed further here). A more extensive survey of all eukaryotes and a transcriptome ORF overlap topology analysis is being done to provide a comparative transcriptomics analysis, similar to the comparative genomics analysis done for prokaryotic ORF overlap topology [57], so will not be discussed further.
A computer program is used to process each mRNA entry with an ORFfinder with three forward frame passes and three reverse complement frame passes. The mRNA entries are filtered to keep only those with at least one ORF ≥ 300 bases in length, where the 3’UTR regions indicated by the ORF’s right boundary are at least 200 bases in length, and begin with a unique 35-base initial 3’UTR sequence for a given prior ORFlength (allows for alt-splice variants to pass). The method only works with operon encoded transcripts if the last ORF in the operon is ≥ 300. Thus it is meant to be applied to genomes with low operon percentage (which favor longer ORFs) to minimize operon recognition failure errors. In the Results is shown how the 3’UTR identification problem occurs in the C. elegans (worm) transcriptome, as anticipated, due to the high operon percentage, and this is found to be the case for axolotl to a smaller extent (not shown), for similar reasons. For the case of the worm and axolotl a simple algorithmic fix is used to reprocess the transcripts passed with ORFs ≥ 300, rescanning their 3’UTRs for ORF ≥ 50, thereby eliminating most of the missed operon structure that is below 300 bases in length that is interfering with the proper delineation of the 3’UTR regions (after the last ORF in the properly identified collection of ORFs in the operon structure). This paper isn’t focused on worm and axolotl, however, so those results are in a separate paper. The software for the ORF overlap topology tabulation is described in the study by Winters-Hilt S (2006) [57], and is briefly summarized in the Supplementary Section-ORFs and ORF overlap topology in prokaryotic dsDNA (Supplementary figures 3-7).
mRNA data for the mouse, tuna, salmon, and worm transcriptomes is examined with six-pass ORF processing: three ORF passes for the different codon framings possible on the ssRNA transcript, and three passes repeated on the reverse complement of the ssRNA sequence. The transcripts with ORF lengths greater than or equal to 300 bases are selected. The ORFs identified from the different ORF passes are then incorporated into a multi track indexing scheme [57], as mentioned in Supplementary Section-ORFs and ORF overlap topology in prokaryotic dsDNA, whereby the ORF overlap topology can be quantified. The non-overlapping ORFs can also be used to ascertain the amount of operon encoding. Once the operon encoding is resolved, the 3’ UTR regions can be identified as the remainder of the transcript after the last ORF of the operon (for a polycistronic transcript), or simply after the (single) ORF in the transcript read to be performed (for a monicistronic transcript). Further selection is then performed at this juncture to restrict to transcripts with 3’ UTR regions with lengths at least 200 bases. Like ORF-length, 3’UTR lengths have heavy-tailed distributions, where the heavy tail regions are where the genes (if ORFs) or miRNA regulatory regions (if 3’UTR) often reside. See table 2 for details. The plot of the length distribution on 3’ UTR regions so identified is given in figure 6, and is used to help guide the choice of the length 200 cut-off to be where the tail of the distribution is entered, where significant deviance from randomness for all events begins to occur.
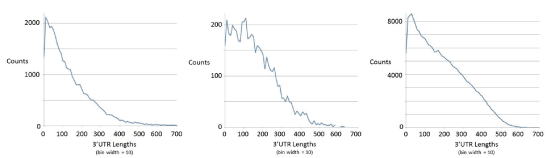
Figure 6: Length distribution on 3’ UTR regions for mouse, tuna, and salmon (from left to right) for the ORFs selected as indicated in table 2.
| Species | #mRNAs (Genbank) |
#ORFs > 300 (and unique 35-length) |
#ORFs (tranlen ≥ 200) |
#Uniq mRNAs |
% mRNAs Dual |
% ORFs Operon |
% ORFs Forward Overlapping |
| Mouse | 98484 | 36654 | 8907 | 7303 | 12.7 | 0.70 | 15.0 |
| Tuna | 10163 | 5366 | 1739 | 1541 | 9.5 | 0.63 | 11.8 |
| Salmon | 498523 | 232014 | 96084 | 82007 | 8.0 | 0.86 | 13.5 |
| Worm | 8327 | 13864 | 8590 | 4660 | 30.4 | 12.9* | ----- |
Table 2: The number of mRNAs used in the transcriptome analysis and their ORF topology characteristics
*Worm result greatly underestimates extent of operon due to ORF ≥ 300 constraint
It is easy to imagine how each strand with ORF ≥ 300 encoding might have a partially overlapping ORF ≥ 300 encoding with different framing (as described in the Background), the percentage of such ORF overlaps in a given transcriptome sample is as shown in table 2 (with accounting for both transcript and reverse complement transcript in the frame-shift overlap analysis). Some ORFs on a given strand do not overlap, necessarily the case if with same global framing, or with separability on other codon framing (from one of the other two codon framing passes). ORFs on the same strand (the transcript sequence ‘as is’ or the reverse complement of the transcript sequence) that do not overlap can be grouped as hypothesized “operons”. This is done in the estimated accounting shown. This results in an upper bound estimate on the true operon percentage, shown in the table 2. It is an estimate since some ORFs may not fit on one operon grouping since it’s only their coding region and small untranslated regions that must fit, i.e., the ORF pieces are generally trimmed on their left ends when it comes to fitting the segments together to have an operon. A quick analysis on ORF track placement for various degrees of left ORF boundary ‘trimming’ allows a means for the operon percentage to be upper bounded, and it is found that the estimates of less than 1% operon structure in the ≥ 300 ORF transcripts are at most 1.5%. This is assuming, however, that the end of any operon structure is being properly identified when the ORFs<300 bases in length are themselves being ignored. From 3’UTR distribution data in figure 6 we see that this is the case for the genomes with low operon structure, which all show very few occurrences of 3’UTR regions greater than 600 bases.
Any missed last ORFs in an operon would greatly add to the length of the 3’UTR region thereby falsely arrived at, and would lead to a distortion in the length distribution of the 3’UTR regions, with the first tell-tale sign of operon recognition failure being in the cutoff on maximum 3’UTR starting to slip to larger values than 600 bases (where other genomes, not shown, also share the trait that 3’UTR regions typically are very rarely greater than 600 bases in length). With worm, however, we expect the operon handling to be insufficient since it is operon rich, which together with the possible occurrence of ORFs < 300 length are now much more significant source of error. The length distribution profile for worm has 3’UTR lengths in significant numbers out to about 1200 bases in length (Figure 7), indicating an operon boundary identification failure due to filtering out ORFs < 300 that are trans to an identified ORF ≥ 300 that is selected in the analysis. For this reason some of the numbers for worm in the table are omitted as entirely invalid, or marked as a lower bound when only providing an estimate of some sort.
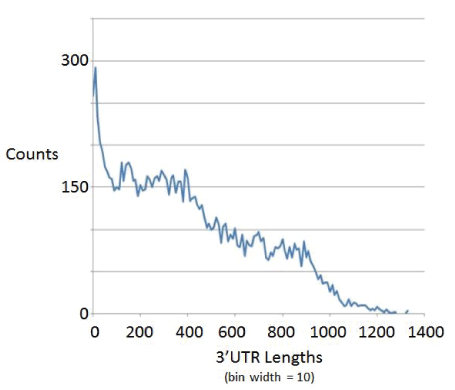
Figure 7: Length distribution on 3’ UTR regions for worm.
The ORF ≥ 300 and 3’UTR ≥ 200 overlapping constructs for the nonoperonic transcriptomes are thus strongly validated as functional given their anomalously long ORF and 3’UTR regions. Consider now that the same analysis has been applied to the reverse complement of the transcript with selection for any coding constructs passing the same stringent cutoffs applied there as well. The transcripts having functional encodings both directly and on their reverse complement are referred to as ‘dually encoded’ in table 2, with the percentage of transcripts with dual encodings between 8% and 13% as shown. Not shown in figure 2 is how similar dual encoding percentages are also seen in a huge variety of fish transcriptomes that are currently under study in a separate effort focused on fish stock diversity assessment (to be described in a separate paper).
The failure of the operon recognition with the worm transcriptome analysis does not directly impact the percentage dual mRNAs similarly revealed, allowing an estimation to be done, and the dual encoding on 3’UTR ≥ 200 transcripts in worm is approximately 30%. Keep in mind this is not a result that would necessarily hold true as a percentage of the entire worm transcriptome (similarly for the other transcriptomes) since there is the restriction to 3’UTR ≥ 200 transcripts. Regardless, whether the amount of dual encoding is 8% or 30% it is still significant and the problem is there is no standard RdRp-like mechanism for accessing the dual genes indicated, or examination of their possible unique disease associations (as a possibly more precariously regulated group). Thus, an active, more significant non-RNAi role for RdRp is hypothesized for eukaryotes. This modifies the central dogma of biomolecular processing (DNAmRNAprotein) to now account for more paths when RdRp is considered (analogous to the extension of the standard model that was made when reverse transcriptase was adopted to allow a path from mRNA back to DNA). Both RdRp and reverse transcriptase are thought to be of viral origin, and the accumulation of both such viral attributes in eukaryotes, but not prokaryotes, leads to a reiteration of the viral eukaryogenesis hypothesis in light of this new information, and this will be discussed in the Discussion and Conclusion sections that follow.
What is remarkable is the appearance of coding-overlap structures in eukaryotic transcripts, with either forward overlapping or, especially, dual overlapping, that are similar to the coding-overlap structures that appear in prokaryotic transcripts (that derive from a dsDNA genome, say, that is dually encoded). It’s as if the endosymbiosis of a viral nucleus left the prokaryote-like genomic information to be ‘lifted’ into the viral-based proto-nucleus over time via their mRNA transcripts, where the reverse complement information is available via RdRp.
As a further validation on the transcripts used in the analysis, that are restricted with selection for ORFs ≥ 300 and 3’UTRs ≥ 200, an analysis of the 7mer motif statistics in the 3’UTR regions is performed (Table 3). The 7mer motif counts are expected to be a richly structured statistic due to the selection pressure from RNAi that uses 7mer miRNA/RISC binding site motifs as loci for RNAi control in the 3’UTR regions. An examination of the 16,384 possible 7mer counts reveals an average count and standard deviation on counts for the various 7mers as shown for mouse, tuna, salmon, and cod. Atlantic cod, has very few high-frequency 7mer structures (less than half that of the others including mouse), strongly indicating a damaged transcriptome for Atlantic cod, which would be consistent with the known overfishing and long-term collapse of the cod fishery in the north Atlantic. For the results in this paper, however, the 7mer results will merely serve to further validate the transcriptome sampling process, especially the very large sample-size Atlantic Salmon data, making the conclusion of significant dual encoding, at the transcriptome level, clear.
Species |
Average nonzero count (µ) |
Standard Deviation (σ) |
o / µ |
Mouse |
162 | 118 | 0.728 |
Tuna |
30.7 | 22.9 | 0.745 |
Salmon |
1821 | 1280 | 0.703 |
Cod |
794 | 919 | 1.157 |
Table 3: 7mer statistical profile validation on 3’UTR
The Discussion begins in the Section-Indications of noninfectious and non-RNAi RdRp role in eukaryotes-new central dogma, with the strong evidence of dual encodings at the transcriptome level and the resulting implications for a noninfectious role for RdRp in eukaryotes. In the Section Viral Eukaryogenesis Hypothesis, the viral eukaryogenesis hypothesis is revisited with refinements according to the long-term role for RdRp that is indicated for eukaryotes. The imprinting of the dual encodings also suggests a pre-existing spliceosome in the viral nucleus. A re-evaluation of the long-term fundamental role of viruses, and other selfish genomic constructs, like transposons and introns as discussed in the study by Winters-Hilt S [34], where the Intron Earliest hypothesis is presented.
The results described in this paper suggest endogenous viral-like RNAdependent RNA polymerase (RdRp) enzymatic processing in eukaryotes at the level of mature mRNA products, such that reverse compliment mRNA transcripts are generated. Once again there appears to be a critical role for certain nucleic acid processing enzymes, such as transposase, when their most common incarnation, or initial discovery, is of viral origin. Such processing could have been introduced via gene transfer from a bacteriophage into a prokaryotic ancestor that eventually branched into the eukaryotic group of organisms. RdRp could have been introduced into eukaryotes, for example, via a viral eukaryogenesis process (as Discussed in the Section-Viral Eukaryogenesis Hypothesis), via gradual evolutionary process whereby viral-infected archaeons/prokaryotes could have been selected for commensalism, then mutualism, and then for an endosymbiotic viral-archaeon/prokaryote relationship involving adoption of the viral type of RdRp.
Endosymbiosis of a membrane-bound organism results in an organelle, as with the mitochondrion or chloroplast, while endosymbiosis of a selfish gene organism (virus or transposon) without encapsulation results in adoption of a gene or selfish genomic construct. The latter case is seen in the many endogenous retroviruses that have been identified, which, if nothing else, offer benefit by providing a competitive inhibition against parasitic exogenous retroviral attacks. What is proposed here is that the endosymbiosis that led to eukaryotic adoption of RdRp could have established the mutualism with a membrane-bound virus that eventually led to a viral endosymbiosis with viral membrane included, the viral membrane then becoming the nuclear membrane of the proto-eukaryotic cell (discussed further in the Section-Viral Eukaryogenesis Hypothesis).
If RdRp is part of a eukaryotic organism’s inheritable complement of biomolecular information processing, then a further modification to the central dogma is needed as shown in figure 8, where more explicit notation of the critical spliceosome stage is shown as well.
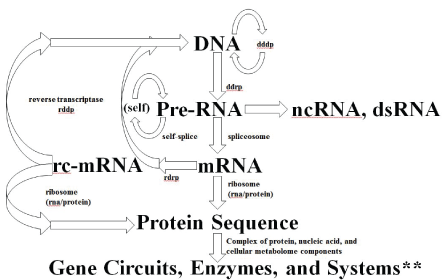
Figure 8: Central Dogma for familiar RNA/DNA/Protein World information processing. Depending on when the development of cellular encapsulation occurs in relation to development of the ribosome and other key enzymes, RNA/DNA/Protein World could have had a pre-cellular phase. Cellular encapsulation could have greatly accelerated the switch (optimization) from ribozyme to enzyme, so once you have a Cell (and virus) you might arrive at the familiar RNA/DNA/ Protein information encoding scheme. The full diagram above might apply to an early universal precursor cell (ancestor to prokaryote and eukaryote), while the same diagram without the last two stages would describe an early universal precursor virus (where the last two stages are implemented for the virus using the host’s cytosol). The selection for more optimized cells and viruses is accelerated via their co-evolution, so co-evolution is itself effectively selected. **By Systems we mean any situation that can give rise to emergent phenomena (or phenotypes) at ‘system’ level, where the role of noise can be one such phenotype.
Given the possibly widespread and thus ancient use of RdRp in eukaryotes, this would indicate that the inception of the proto-eukaryotic cell line would have been marked by the adoption of a native RdRp capability. Consider also that at the inception of the eukaryotic cell line there is the adoption of a nucleus (by definition) with the possible wholesale adoption of a spliceosomal machine protected by the nuclear membrane, as well as any other nucleic acid processing that might be protected by that membrane boundary. For the proto-eukaryote there is also thought to be a more energy-rich metabolic suite of capabilities than is typical for prokaryotes. For this reason the archaeon/prokaryotic-like organism involved in the viral eukaryogenesis may have already adopted a proto-mitochondrial endosymbiont, allowing it to perform significantly more ATP production at the mitochondrial membrane, not the cellular membrane, freeing the cellular membrane to become simpler (e.g., no wall, just a single membrane) allowing greater cellular communication (and greater susceptibility to membrane-bound virus invasion). Now consider the archaeon/prokaryotic-like cell scenario in the co-evolutionary context of an ancient evolutionary battle between virus and cell. As the archaeon/ prokaryotic-like cell develops more and more refined RNA interference defenses it forces the co-evolving virus to compensate, by learning how to co-opt the those defenses for its own use, and by adoption of a membrane boundary for invasion by internalization and operation as an internal (nuclear) boundary (in some cases, such as for poxviruses [39]) to shield its RNA processing from interference. All that’s needed for the host virus system to move towards mutualism is a trade-off, such as the host getting the use of RdRp, and where the virus gets the usual access to the host cellular machinery. Viral eukaryogenesis is, thus, hypothesized in this picture as the simplest description given the organisms and their interplay that was involved at the time.
As mentioned in the Results, there is a remarkable ‘fingerprint’ from the prokaryotic-like precursor’s genome/transcriptome in the eukaryotic transcriptomes we see today. This is because the retention of the archaeon/ prokaryotic-like genomic information seems to be by way of adoption of the archaeon/prokaryotic-like organism’s transcripts, where the transcript that carried a particular gene with an overlap encoding is adopted, with overlap information intact, into the proto-eukaryotic transcriptome processing, and eventually (possibly via reverse transcriptase) gets written into the viral/nuclear dsDNA genome (assuming a dsDNA virus). Thus, the highly compact overlap, operonic, and dual encoding found for prokaryotic genes is found imprinted not via genome-genome transfer, but via transcriptome-genome transfer. Given the splicing to arrive at transcript; however, these results in the transcript only producing genomic information from one strand, with the other (dual) information necessarily accessed via RdRp. With the viral mutualism hypothesized already in-place, however, this accessibility of RdRp would have not been a problem.
There are a number of assumptions leading up the specific forms of viral eukaryogenesis hypothesis outlined above. Part of the ‘stage that is set’ involves the proto-archaeon/prokaryotic cell and ancient virus co-evolution in place at the time. Whether viruses had spliceosome processing already (and it was ancient) or whether nucleic acid splicing was a remarkably late invention, this is another assumption that impacts the viral eukaryogeneis model. In the study by Winters-Hilt S [34], the Intron Earliest hypothesis is posed, such that the viruses arrived at the viral eukaryogenesis with splicesome already present, this being one of the selection pressures for them to protect against RNA interference by adoption of membrane enclosure in their ‘trojan horse’ endocytosis attack (that eventually leads to endosymbiosis when entering a mutualism relation). In the Intron Earliest hypothesis, the spliceosome processing is ancient and carried in the viral line since selection pressure on proto-prokaryotes led to loss of introns (and loss of the need for the spliceosome). RdRp is similarly thought to be ancient and carried in viral line and similarly lost in proto-prokaryotic line as shift to DNA and larger genomes (and host complexity) led to shift to DdRp in cells.
The viral eukaryogenesis theory also suggests a possible origin for meiosis and sex [56], where it is proposed that the mitotic cycle evolved from virus established with a permanent lysogenic presence, and the meiotic cycle (and sex) evolved from the process whereby the virus transferred to new hosts. Also in the study by Bell P [56] was discussed the process whereby the viral-nucleus dominated cellular control led to a reorganization of the prokaryotic transcription/translation regime into the typical eukaryotic process whereby mRNA is capped prior to extrusion into the cytoplasm, and where the cap binding protein directs translation of the capped mRNA. Reorganization covers an evolutionary refinement process in the proto-eukaryote where non-nuclear genomic information gets ‘lifted’ into the nucleus. Perhaps the same proteins that participate in the spliceosomal activity in the nucleus, bind nucleic acid in cytosol on their way from their cytosol production and assembly into the nucleus [30,31], allowing reverse transport, with reverse splicing of introns already occasionally occurring inside the nucleus, reverse transcriptase (RT) would then be all that’s needed to pull the ‘intronified’ transcript information permanently into the viral/nuclear genome. This could easily occur since RTs are a common component of viruses, even the simplest viruses. In the evolutionary lift procedure outlined, an archaeon/prokaryotic host transcript would be mapped to an ‘intronified’ viral-nucleus genome sequence. This is hypothesized to have occurred given the distinctive statistical artifact that is seen in the Results-e.g., eukaryotic mRNA transcripts are seen with overlap encoding information with respect to their reverse complement reads (tracing back to noneukaryotic mRNA transcripts with such overlap encoding, which then relates directly to the same overlap encoding at the non-eukaryotic intronless dsDNA genomic level). The Results, thus, support a form of the viral eukaryogenesis hypothesis with a genomic uptake via ‘intronification’, further suggesting that the viral nucleus already had control of the spliceosomal machinery. The Results also suggest a possibly larger role for RdRp than purely RNAi-related
An analysis of mRNA data reveals that mRNA transcripts passing stringent validation conditions, having at least one ORF with length ≥ 300 nucleotides in length and with 3’UTR length ≥ 200 nucleotides, have a significant amount of overlapping reverse complement encoding structure passing similar stringency tests. The overlap encoding revealed for the ssRNA is what might result if a reverse complement mRNA could be generated, such as by RNA-dependent RNA polymerase (RdRp), and is analogous to the overlap encoding that might exist on a prokaryotic dsDNA genome. This is indicative of three things: (i) RdRp might play a larger role in eukaryotes than to support RNAi, with associated changes to the central dogma; (ii) ancient remnants of the imprinting of an archaeon/prokaryotic overlap encoding at the genome/transcriptome level appear on the proto-eukaryotic transcriptome with resulting overlap encoding at transcriptome level in eukaryotes; (iii) a specific form of viral eukaryogenesis hypothesis is suggested, where the viral ancestor provided both RdRp and the spliceosome.
The author would like to thank Connecticut College for research support.
Download Supplementary Data Here
Download Provisional PDF Here
Article Type: Research Article
Citation: Winters-Hilt S (2017) RNA-Dependent RNA Polymerase Encoding Artifacts in Eukaryotic Transcriptomes. Int J Mol Genet Gene Ther 2(1): doi http://dx.doi.org/10.16966/2471-4968.108
Copyright: © 2017 Winters-Hilt S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access