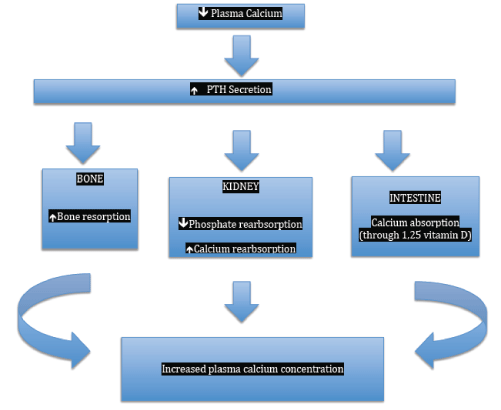
Figure 1: Physiological effects of parathyroid hormones.

Ernest Yorke* Yacoba Atiase Josephine Akpalu
Endocrine and Diabetes Unit, Department of Medicine and Therapeutics, University of Ghana, Ghana*Corresponding authors: Ernest Yorke, Endocrine and Diabetes Unit, Department of Medicine and Therapeutics, University of Ghana, Ghana, Tel: +233 206301107; E-mail: pavlovium@yahoo.com
Primary Hyperparathyroidism (pHPTH) is one of the common endocrine conditions usually affecting women above 45 years and it is characterised by hypercalcaemia with associated inappropriately raised circulating or normal Parathyroid Hormone (PTH). Since the widespread use of multichannel auto analysers from the beginning of the 1970s, the incidence of primary hyperparathyroidism has been rising globally. In areas where this is routinely done, the disease is being diagnosed early and usually asymptomatic; compared to areas with less routine screening like Africa, the disease usually presents with skeletal complications or kidney stone disease. This review presents an update and highlights the nuances of diagnosis as well as variants of primary hyperparathyroidism, screening for complications, localisation of tumour, treatment of symptomatic disease and asymptomatic disease, surgical approaches including post: surgical care as well as non: surgical/medical approaches. We further highlight areas of controversy and future research such as the pathophysiology and natural history of Normocalcaemic Primary Hyperparathyroidism (NPH) as well as monitoring and indications for surgery for asymptomatic disease.
Parathyroid gland; Parathormone; Hypercalcaemia; Complications
ALP: Alkaline Phosphatase; BMD: Bone Mineral Density; CaSR: Calcium Sensing Receptor; CDC: Cell Division Cycle; CDNK: CyclinDependent Kinase Inhibitor; CT: Computer Tomography; DEXA: Dual Energy X-Ray Absorptiometry; ECF: Extracellular Fluid; eGFR: Estimated Glomerular Filtration Rate; EMR: Electronic Medical Record; ENT: Ear, Nose and Throat; FHH: Familial Hypocalciuric Hypercalcaemia; FIHPT: Familial Isolated Hyperparathyroidism; GU: Genitourinary; HPT-JT: Hyperparathyroid-Jaw Tumour; MEN: Multiple Endocrine Neoplasia Syndromes; MRI: Magnetic Resonance Imaging; NPH: Normocalcaemic Primary Hyperparathyroidism; NSHPT: Neonatal Severe Hyperparathyroidism; pHPTH: Primary Hyperparathyroidism; PTH: Parathyroid Hormone/Parathormone; PTHrP: PTH-Related Peptide; SPECT: Single Photon Emission Computed Tomography; Tc: Technetium; TSH: Thyroid Stimulating Hormone; USA: United States of America; VFA: Vertebral Fracture Assessment
Hyperparathyroidism may be primary, secondary or tertiary. In primary hyperparathyroidism, there is hypercalcaemia associated with inappropriately raised circulating or normal PTH. This may result from a single adenoma (85%), multiple adenomas or hyperplasia (both constituting about 15%) and rarely from parathyroid carcinoma [1]. Parathyroid cancers tend to be large tumours with evidence of invasion of surrounding structures; however, in some cases parathyroid cancers may are appear as benign tumours by being completely encapsulated. The most common sites for ectopic adenomas are within the thyroid gland, the superior mediastinum, and within the thymus, and occasionally in the retroesophageal space, the pharynx, the lateral neck, and even the submucosa of the oesophagus.
Secondary hyperparathyroidism occurs where appropriately raised PTH in response to chronic hypocalcaemia with resultant hyperplasia usually from chronic renal failure [2] and occasionally from Vitamin D deficiency. When secondary hyperparathyroidism persists, the parathyroid gland may become autonomous resulting in hypercalcaemia associated with inappropriately raised PTH, i.e., tertiary hyperparathyroidism.
Recently, a newer phenotype where there is raised PTH but normal calcium (called Normocalcaemic Primary Hyperparathyroidism) is been recognized and is described in more detail later [3].
Primary Hyperparathyroidism (pHPTH) is one of the common endocrine conditions with a female preponderance (female:males ratio 3:1) [4,5] usually affecting women above 45 years. Since the widespread use of multichannel auto analysers from the beginning of the 1970s, the incidence of primary hyperparathyroidism has been rising [6]. Studies done in Mayo Clinic, United States of America (USA) revealed a 4-5 folds increase in the incidence of primary hyperparathyroidism to approximately 100,000 new cases per year or about 22 cases per 100,000 person years [4,6]. In another study by Yeh, et al. [5] done from 1995-2010, a period well after routine biochemical screening available in United States of America (USA), the prevalence of primary hyperparathyroidism has been found to have increased by three-fold. Data from Europe suggests a prevalence of about 1-7 per 1000 adults [7-9]. However, in contrast, a population-based data from Olmsted County, Minnesota, in the USA indicate that the prevalence of primary hyperparathyroidism is decreasing slowly [4]. In terms of racial differences in prevalence, primary hyperparathyroidism was found to be more common in African Americans followed by Caucasians and Asians [10].
Whilst the aetiology of primary hyperparathyroidism is not clear, most patients with primary hyperparathyroidism have sporadic disease. Familial clusters may occur (Table 1) in multiple endocrine neoplasia syndromes (MEN 1 or MEN 2A), Familial hypocalciuric hypercalcaemia (FHH), Hyperparathyroid-Jaw tumour (HPT-JT) Syndrome, Familial isolated hyperparathyroidism (FIHPT) and Neonatal severe hyperparathyroidism (NSHPT) [11]. The genetic basis of MEN 1 and MEN 2A are an inactivating mutation of MEN 1 gene (which is located on chromosome 11) and a germline mutation of Ret proto-oncogen located on chromosome 10, respectively [11]. In HPT-JT, Germline mutation of HRPT2 localized on chromosome arm 1q is responsible whilst in the case of FIHP, there is heterogeneous genetic makeup. FHH is an autosomal dominant condition where there is heterozygous loss of function from calcium sensing receptor (CaSR) mutations. Individuals who are homozygous for CaSR mutations develop neonatal severe hyperparathyroidism [12]. The lack of responsiveness of parathyroid hormone to extracellular calcium is mainly due to altered sensitivity of these abnormal clonal parathyroid cells and partly due to the sheer increase in parathormone production from the increases numbers of these cells. The results of these changes is that for a given extracellular calcium concentration, PTH is higher [13].
| Syndrome | Mechanism | Features |
| Multiple endocrine neoplasia type 1 | Inactivating mutation of MEN1 gene (chromosome 11) | Parathyroid, anterior pituitary and pancreas tumours |
| Multiple endocrine type 2A | Germline mutation of Ret proto-oncogen (chromosome 10) | Pheochromocytoma, medullary thyroid cancer and cutaneous lichen amyloidosis |
| Familial hypocalciuric hypercalcaemia | Heterozygous loss of function calcium sensing receptor (CaSR)/mutations | Hypocalciuria, rare pancreatitis |
| Neonatal severe hyperparathyroidism | Homozygous loss of function calcium sensing receptor (CaSR)/mutations | Life-threatening hypercalcaemia and hypotonia |
| Hyperparathyroidism-jaw tumour syndrome | Germline mutation of HRPT2/CDC73 (chromosome 1q) | Mandible/maxillary fibromas, parathyroid cancer, uterine and renal tumours |
| Familial isolated hyperparathyroidism | Heterogenous e.g MEN 1, CASR, HRPT2, CDKN1B | Non-specific features |
Table 1: Familial Syndromes Associated with Primary Hyperparathyroidism [11,12,14].
MEN 1-Multiple endocrine neoplasia type 1; CaSR- Calcium sensing receptor; CDC- Cell division cycle; CDNK- Cyclin-dependent kinase inhibitor
Extracellular calcium concentration is mainly maintained by parathyroid hormone or parathormone (PTH) secreted by the parathyroid glands; and secretion of PTH is influenced by ionized calcium. The chief cells of the parathyroid glands secrete PTH as a polypeptide containing 84 amino acids, which is a prohormone; its activity depends on effective hormone-receptor interaction of the 34-N-terminal amino acids part [15]. Most people have 4 parathyroid glands situated on the posterior aspects of the thyroid gland and are described as inferior, superior, right or left depending on their position [16]. Occasionally 3, 5, or, occasionally, more glands may be found in some people [17]. Embryonically, the superior parathyroid glands are derived from the fourth pharyngeal pouch, a structure that also give rise to parafollicular or C cells of the thyroid gland; the superior glands may thus be found within the substance of the thyroid gland because of their common origin [16]. The inferior glands are derived from the third pharyngeal pouch, which is also the embryologic origin of the thymus, and are usually found close to or within it [16].
The primary function of PTH is to maintain the Extracellular Fluid (ECF) calcium concentration within a narrow normal range through a feedback mechanism between ionized calcium and PTH (Figure 1). Calcium-Sensing Receptors (CaSR) in the parathyroid glands are activated when calcium levels drop below 2.5 mmol/L (10 mg/dl), which in turn stimulate the release of PTH, reaching a peak when calcium falls below 1.8 mmol/L (7.5 mg/dl) [18]. At a calcium level of above 2.5 mmol/L (10 mg/dl), the secretion of PTH is minimal. PTH actions include increased release of calcium and phosphate from bone. It also activates the 1-alpha-hydroxylase enzyme of renal proximal tubules which in turn converts 25-hydroxyvitamin D into the more active 1,25-dihydroxyvitamin D-3 (calcitriol) which in turn promotes the reasborption of calcium and phosphate at the intestinal level. PTH increases renal (thick ascending limb of the loop of Henle and distal convoluted tubule) reabsorption of calcium and promoting phosphaturia [19]. Overall, these actions of PTH increase serum calcium concentration. Apart calcium, which is the main agonist of the CaSR of the parathyroids, magnesium (Mg) and other cations activate the CaSRs to influence parathyroid release and function. Magnesium modulates PTH secretion in a way similar to calcium [20-22]. However, high levels and severe decrease in serum Mg alter adenylate cyclase activity and production of Cyclic Adenosine Monophosphate (cAMP) leading to impaired PTH release and function [22-24].
Figure 1: Physiological effects of parathyroid hormones.
Some malignancies including cancers of the lung and sometimes of the breast, head, neck, bladder, gastrointestinal tract, and ovaries, as well as leukaemia and lymphoma may secrete PTH-Related Peptide (PTHrP) [25]. It has the same N-terminal as PTH, and therefore it can bind to the same receptor and stimulate the action of PTH leading to humoral hypercalcaemia of malignancy [25]. PTHrP-associated hypercalcaemia is far more common than the production of ‘true’ but rare PTH produced by non-parathyroid malignancies (ectopic PTH) [26-28]. Indeed, it is the most common cause of hypercalcaemia in hospitalized patients [25,29]. The hypercalcaemia of malignancy due to PTHrP results in a low or undetectable intact parathormone levels. PTHrP may also be monitored as a tumour marker during the treatment phase of the underlying malignant disease and PTHrP does not seem to have a major influence on the production of 1,25-dihydroxycholecalciferol [25].
Single and multiglandular diseases are indistinguishable in terms of clinical symptoms. However, younger patients (<40 years), and particularly those with a personal or family history of a Multiple Endocrine Neoplasia (MEN) syndrome, are more likely to have multiglandular disease.
The effect of chronic hypersecretion of PTH from primary hyperparathyroidism may result in osteopenia from increased bone resorption, and in severe cases, osteitis fibrosa cystica, a condition, which is hardly seen in clinical practice currently but characterized by subperiosteal resorption of the distal phalanges, tapering of the distal clavicles, salt-and-pepper appearance of the skull, and brown tumours of the long bones. With advent of automated chemistry analyzers, radiological manifestations of primary hyperparathyroidism have fallen from 23% in the series by Cope [30] to less than 2% by the series of Wass, et al and Silverberg, et al. [31,32]. Due to this fact, overt skeletal disease in primary hyperparathyroidism skeletal X-ray may not be indicated during evaluation for primary hyperparathyroidism.
Others include renal calculi from increased calcuria, polyuria and dehydration, muscle weakness, abdominal pains and occasionally hypertension. Hypercalcaemia may be associated with peptic ulcer disease (from increase gastric acid secretion) as well as pancreatitis. Neuropsychiatric manifestations may include depression, confusion and personality changes. In the majority of cases however, renal and skeletal complications are not demonstrable [3].
The clinical presentation in less developed or poorer countries where Vitamin D deficiency is commonplace, and also where screening tests for biochemical abnormalities are less frequently done, tend to be more severe with classical features such as osteitis fibrosa cystica as well as other skeletal and renal complication. [33] In the presence of Vitamin D deficiency, primary hyperparathyroidism presents with severe phenotypes [34] such as with larger adenomas, higher serum parathyroid hormone, serum calcium and alkaline phosphatase levels as well as lower bone mineral density, and higher rates of fractures [35-40] (Table 2).
| System involved | Symptomatology |
| Neuropsychiatric | Behavioural disturbances
|
| Gastrointestinal | Anorexia Constipation Nausea and vomiting Pancreatitis Peptic ulcer disease |
| Renal | Nephrogenic diabetes insipidus Nephrolithiasis Tubular dysfunction Chronic renal failure |
| Cardiovascular | Shortened QT interval on electrocardiogram ST-segment elevation mimic Hypertension Ventricular arrhrythmias |
Table 2: Clinical Presentation of Primary Hyperparathyroidism [41-50].
The presentation of parathyroid cancers, which form less than 0.5% of all primary hyperparathyroidism cases, is quite different. In some series, however, an incidence as high as 5% has been reported [51,52]. In contrast to benign parathyroid disease (Table 3), where there are more females compared to men, parathyroid cancer occurs with equal frequency in both sexes. Its presentation typically runs an indolent yet progressive course and coupled with the fact that the histology of parathyroid tumours can be equivocal or misleading, the diagnosis of malignancy is often made only when local recurrence or metastases occur [53]. Tissue invasion and local or distant metastases appear to be the key indicators of malignancy. On histology, mitotic activity and nuclear atypia are not sufficient by themselves to make the diagnosis of parathyroid cancer.
| Characteristic | Benign | Malignant |
| Sex ratio (female: male) | 3-4:1 | 1:1 |
| Average age (years) | 55 | 48 |
| Serum calcium (mmol/L) |
Less or equal to 2.8 | >3.5 |
| Serum PTH | Mildly elevated | Markedly elevated |
| Cervical mass, palpable | Rare | Common |
| Bone and renal involvement | Rare | Common |
| Size and appearance of tumour | Smaller, dark brown, and firm but not hard | >3 cm and may be palpable; hard and gray- white and adherent to adjacent structures |
Table 3: Useful distinguishing features between benign and malignant primary hyperparathyroidism [52].
Parathyroid cancers tend to have very high levels of serum calcium (usually above 3.5 mmol/L), markedly raised parathyroid hormone (up to 20 fold increase) with almost every affected patient being symptomatic; non-functional parathyroid cancers are extremely rare. It may also present with a neck mass, bone and stone disease. Renal involvement such as nephrocalcinosis, nephrolithiasis and impaired renal function occur in up to 80% of patients, and bone involvement (osteitis fibrosa cystica, subperiosteal resorption, “salt and pepper” skull, and diffuse osteopenia) in up to 90% [52]. Renal colic is a frequent presenting complaint whilst other symptoms include muscle weakness, fatigue, depression, nausea, polydipsia and polyuria, bone pain, fractures, recurrent severe pancreatitis, peptic ulcer disease, and anaemia.
Parathyroid cancer usually occur as a sporadic disease but may be associated with hereditary syndromes of hyperparathyroidism [54,55] particularly hyperparathyroid-jaw tumour syndrome [56], a rare autosomal disorder, in which as many as 15% of patients will have malignant parathyroid disease. On rare occasions, parathyroid carcinoma has been reported in patients with longstanding secondary hyperparathyroidism [57,58]. Other characteristics of parathyroid cancers include younger age at onset (40-50 years) compared to benign tumours (50-60 years). Parathyroid cancers have also been referred to as cystic parathyroid adenomatosis because cystic changes are common [59,60].
The diagnosis of primary hyperparathyroidism is made by demonstrating raised serum calcium associated with inappropriately elevated or normal intact PTH [61]. Apart from a parathyroid adenoma or cancer, other differential diagnosis must be considered. The presence of raised 24-hour urinary calcium excretion rules out familial hypocalcuric hypercalcamia where there is hypocalucuria [62]. Familial hypocalciuric hypercalcaemia, an autosomal dominant disorder renal calcium-sensing receptor, should be considered in patients with long-standing hypercalcaemia, urinary calcium levels less than 100 mg/24 hours, and calcium to creatinine clearance ratio less than 0.01 on normal calcium diet [63]. The inactivating mutation of the Calcium sensing receptor (CASR) gene results in an increase in the set point for serum calcium suppression of PTH secretion [62]. Because of the asymptomatic nature of the hypercalcaemia, surgery is not indicated in its management. It is possible to request CASR gene sequence testing when on the diagnosis is in doubt [27].
Lithium therapy [64] and thiazide use [65] may result in a similar serum biochemical picture. Long-term lithium therapy decreases sensitivity to calcium within the parathyroid gland and may also reduce urinary calcium excretion, however, short-term use may be associated with reversible hypercalcaemia [64,66]. Whilst it may sound reasonable to discontinue lithium therapy in these circumstances, it is not always easy to substitute lithium. Lithium-associated primary hyperparathyroidism occurs in up to 15% of long-term users [67]. In vivo studies suggest that lithium stimulates PTH secretion directly [68,69] and also affects calcium metabolism at the renal level and accelerates loss of bone density [67,70]. In practice lithium has been associated with causing more parathyroid adenomas rather than hyperplasia [67]. It has been postulated that lithium serves as a mitogen in causing adenoma or enhancing the growth of a previously unrecognized abnormal parathyroid tissue [8,64].
The hypercalcaemia associated with thiazide use is presumed to be multifactorial [71,72] and most hypercalcaemic patients taking thiazide diuretics will eventually be diagnosed with primary hyperparathyroidism [73]. Thiazide- induced enlargement of the parathyroid glands may or may not be demonstrated [74-75]. This pathophysiological factor for thiazide induced hypercalcaemia include thiazide-associated reduced urinary calcium excretion, a pH-dependent increase in protein-bound calcium resulting from a metabolic alkalosis, and increased intestinal calcium absorption [71,72]. It is also thought that thiazides might unmask mild or normocalcemic primary hyperparathyroidism [76]. If there is no contraindication for its continuation, thiazide therapy could be discontinued when there is thiazide associated primary hyperparathyroidism. A re-evaluation of the calcium and parathormone levels in 3 to 6 months may reasonable.
Albumin remains the major calcium binding protein and alterations in their concentration affect measured calcium; and therefore total serum calcium must be adjusted to reflect any abnormality in albumin. The formula usually used is:
Corrected calcium (mmol/L)=Measured total serum calcium in mmol/L+0.20 × (4.0-patient’s serum albumin concentration in g/L)
Free calcium concentration measured by ionised calcium concentration is increasingly becoming available in many centres and can be used if available.
The measurement of ‘intact’ PTH helps distinguish between primary hyperparathyroidism and malignancy-associated hypercalcaemia, the next most common cause of hypercalcaemia. Except in the case of FHH, thiazide diuretics, lithium therapy, malignancy-associated hypercalcaemia, in all causes of hypercalcaemia, PTH is suppressed. On occasion, the PTH levels will be in the upper range of the normal value 50. Parathyroid hormone-related protein (PTHrP) is responsible for the hypercalcaemia in malignancy-associated hypercalcaemia.
Typically, the reference range for second generation ‘intact’ PTH is as 10-65 pg/ml. Because PTH levels normally rise with age, in individuals below the age of 45, the upper limit of normal for PTH should be taken to be closer to 45 pg/mL. There exists a third generation assay which measures ‘whole’ PTH because it is more specific for the 1-84 molecules [77]. Because of this property, cross-reactivity with large amino terminal truncated fragments does not occur. However, this assay detects a post-translational form of PTH constituting about 10% of measured PTH and tends to be higher in renal failure, parathyroid cancer and severe hyperparathyroidism [78]. The normal range for the third generation assay for PTH is typically about half that of the second-generation assay (5-35 pg/mL). However, overtime, the diagnostic sensitivities of ‘whole’ and intact assays for most clinical purposes appear to be similar with the second-generation assay being more widely used [50]. In the use of these two modern assays described above, PTH and parathyroid related hormone (PTHrP) do not cross react.
Recently, there has been the recognition of normocalcaemic primary hyperparathyroidism (NPH), a condition where total and ionized serum calcium are completely normal but in whom the PTH level is persistently elevated [76]. It is suggested to be a variant of primary hyperparathyroidism. [76,79-82] It is important to exclude all causes of secondary hyperparathyroidism including Vitamin D deficiency in order to make the diagnosis of NPH. Once Vitamin D have been replaced when there is deficiency, and other possible causes of elevated PTH such as renal failure has been excluded, yet serum calcium remain normal, the diagnosis of NPH should be considered strongly. Patients with normal calcium levels and elevated parathyroid hormone levels in the absence of an identifiable secondary cause should be monitored for progression to hypercalcaemia [50]. In large population based study where over 5,000 post menopausal had baseline screening and repeated after 8 years, it was observed that many patients developed hypercalcaemia [83,84]. In an observational study by Lowe, et al. [79] where 37 patients were followed for a mean of 3 years, hypercalcaemic primary hyperparathyroidism occurred in 7 (19%) individuals. Also, 40% developed evidence of disease progression such as kidney stones, fractures, marked hypercalciuria and a reduction in Bone Mineral Density (BMD) of more than 10%. Of note, 7 patients had successful parathyroidectomy [79]. Amongst unreferred and non-selected population where parathyroid disease was not being sought, NPH prevalence rates ranged between 0.4% and 3.1% [33]. These figures were supported by the results when the Electronic medical record (EMR) of a large, tertiary referral centre was examined to study the prevalence of undiagnosed and unrecognized primary hyperparathyroidism [85].
For the first time in 2014, NPH was recognized as a form of primary hyperparathyroidism at the 4th International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism, where it also suggested guidelines (Figure 2) for its management [86]. Such patients should have annual serum calcium and PTH measurements done and a DEXA scan every 1-2 years. The conference suggested that if hypercalcaemia develops established guidelines for the management of primary hyperparathyroidism applies. In situations where hypercalcaemia does not develop but rather classical complications of primary hyperparathyroidism, such as kidney stones, reduced bone mineral density, fractures, parathyroidectomy is recommended [86].
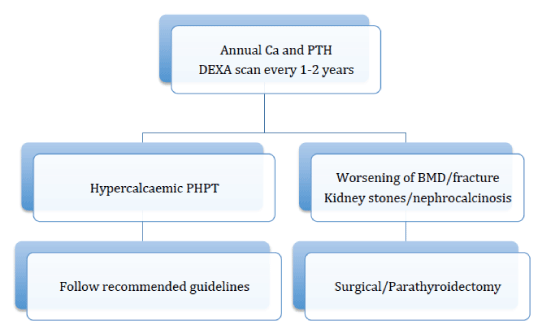
Figure 2: Algorithm for managing normocalcaemic primary hyperparathyroidism [86].
In some cases of primary hyperparathyroidism patients may occasionally have normal total and/or ionized calcium, though they are hypercalcaemic most of the time. In such instances, repeated calcium measurements may be warranted [78].
Nephrolithiasis still represents the most common complication of primary hyperparathyroidism despite the fact that this incidence has fallen from about 60% to 20% in line with increases automated measurement of serum calcium [30,61,87] which tend to pick up asymptomatic hypercalcaemia early. A recent study by Cipriani, et al. [88] reported that 55% of patients with primary hyperparathyroidism had kidney stones by abdominal ultrasound. Consequently, renal imaging using ultrasonography, CT scan or other modality, should be performed in primary hyperparathyroidism [50].
The reason for the increased stone formation in primary hyperparathyroidism is thought to be multifactorial [89,90]. Urinary calcium excretion is not thought to be a highly rated factor for kidney stones in primary hyperparathyroidism [91], but along with other urinary risk factors are helpful predictive indices [86]. Scillitani, et al. [89] suggests that specific polymorphisms of the calcium receptor gene might be an important factor in the pathogenesis of kidney stones.
Reduction in creatinine clearance has been associated with primary hyperparathyroidism; however, the reason for this is unclear. PTH levels tend to rise when the eGFR value is less than 60 ml/ min/1.73 m2 , in the absence of primary hyperparathyroidism. The Hyperparathyroidism Workshop Panel [86] has therefore set this value as a level for recognizing renal impairment. However, this associated reduction in creatinine clearance was not found in a recent study where PTH levels were similar among individuals with creatinine clearance is 30-60 ml/min compared to those with clearances greater than 60 ml/min [92]; though at the histological structure level, those with creatinine clearance <60 cc/min showed more active disease [93].
During the proceedings of the Fourth International Workshop on asymptomatic primary hyperparathyroidism, it was observed that primary hyperparathyroidism causes site-specific loss of bone mineral density (BMD) and may predispose patients to fragility fractures. Consequently a DEXA scan should be considered for all patients with primary hyperparathyroidism to screen for clinically relevant skeletal manifestations [94]. Its effect is mainly on cortical bone. The use of DEXA scan in assessing for bone mineral density should be measured at the lumbar spine, hip, and distal radius [63]. Some studies have shown that fracture incidence increases in the forearm but reduced, or at least unchanged, in the lumbar spine as compared to control subjects without primary hyperparathyroidism [95-97]. Other studies also rather reported increases in vertebral fractures [98,99]. A closer look at the study by Khlosa, et al. [98] where they did a retrospective assessment of fracture incidence over a 28-years period, found out that fracture rate was increased not only at the forearm but also at central, vertebral sites. Another study also reported an increase in vertebral fractures in postmenopausal women with primary hyperparathyroidism [100].
A positive family history of parathyroid tumours may warrant genetic testing. MEN types 1 and 2A, hyperparathyroidism jaw tumour syndrome, neonatal severe primary hyperparathyroidism, and familial isolated hyperparathyroidism are the most characterized hereditary syndromes [14,101]. Prior history of ionizing radiation to the head and neck has been recognized as being associated with the development of primary hyperparathyroidism decades after the initial exposure [102-104].
Imaging of the parathyroid gland is not needed for confirmation or the decision to do surgery. It helps in appropriate planning for surgery [67]. Localisation techniques include ultrasound, CT scanning, positron emission tomography and Sestamibi scanning. Ultrasound is the most available and cheapest imaging modality with a sensitivity range from 42-82% and specificity of approximately 90% [105]. Ultrasound imaging is non-invasive and allows concomitant thyroid pathology to be detected and results can be integrated with Sestamibi scanning [106]. CT scanning can apart from assessing the anatomy of the parathyroid gland can also be useful for detecting ectopic parathyroid tissue the mediastinum [107]. Four dimensional CT Scans is increasingly becoming a preferred localization approach in a number of centres because it provides additional information which guides the surgical approach [108]. In cases where there is a mediastinal tissue and in individuals who have persistent disease following parathyroidectomy, MRI with contrast may be useful [109].
Sestamibi is most useful for identifying single adenomas and ectopic PTH tissue as may be found in the mediastinum. Whilst the sensitivity may vary among institutions, with experience in its use, it has a sensitivity of about 90%. Localization can be improved with a combination of Sestamibi with single photon emission computed tomography (SPECT) alone [110] or in combination with CT scan [111]. When there is persistent, recurrent or invasive disease, invasive techniques such as arteriography and selective venous sampling may be useful to localize the lesion [112]. This is particularly useful when other imaging modalities have not been able to identify the abnormal parathyroid tissue.
Surgery is required for all patients with typical parathyroidrelated symptoms involving skeletal, renal or gastrointestinal systems; history of an episode of life-threatening hypercalcaemia; significant neuromuscular or psychological symptoms without other obvious cause [50,63]. The American Association of Endocrine Surgeons Guidelines for Definitive Management of Primary Hyperparathyroidism also recommends strongly parathyroidectomy for patients with neurocognitive and/or neuropsychiatric symptoms that are attributable to primary hyperparathyroidism, although, the evidence for this recommendation is weak [63].
The surest way for successful treatment is by surgery with removal of the abnormal tissue by an experienced surgeon [113]. Surgical management is cost-effective than long-term observation or pharmacologic therapy [63,114]. Approaches to surgery are discussed in full detail later.
With the increasing use of automated chemistry analysers, most people with primary hyperparathyroidism have asymptomatic disease without symptoms or signs that are usually linked with the associated hypercalcaemia or excess parathormone.
Despite the fact that surgery remains the only definitive mode of cure, the preponderance of asymptomatic cases has led to a lack of clear consensus as to whether the right mode of treatment for patients should be surgery for this group of patients. The argument for surgery is that mild diseases will progress over time [115].
The Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism issued some guidelines for the management of asymptomatic primary hyperparathyroidism in 2014 [86]. In the guidelines (Table 4), they recommend that among asymptomatic patients with hyperparathyroidism, surgery may be considered when any of the following is present: serum calcium concentration more than 0.25 mmol/L (1 mg/dL) above the upper limit of normal; BMD by DEXA T-score<-2.5 at lumbar spine, total hip, femoral neck, or distal 1/3 radius; vertebral fracture by X-ray, CT, MRI, or Vertebral Fracture Assessment (VFA); estimated glomerular filtration rate (eGFR)<60 cc/min; 24-hour urine for calcium; >400 mg/ day (>10 mmol/day) and increased stone risk by biochemical stone risk analysis; presence of nephrolithiasis or nephrocalcinoisis by X-ray, ultrasound, or CT scan; and Age <50 years. Surgery may also be recommended for those who do not qualify for surgery but are unwilling to comply with follow up protocols or circumstances may not allow such as long distances from the treatment centres [86].
| Indication | Specific criteria for Surgerya |
| Skeletal | |
| Bone Mineral Density | b,cT-score of -2.5 or less at the lumbar spine, femoral neck, total hip, or distal 1/3 radius by DEXA |
| Fracture | Vertebral fracture identified by X-ray or VFA, even if there is no prior documentation |
| Renal | |
| Creatinine clearance | Creatinine clearance of <60 cc/min |
| Renal stone | Renal imaging with X-ray, ultrasound, or CT scan identifying stones or nephrocalcinosis |
| 24-hour urinary calcium | Marked hypercalciuria ( >400 mg/d) in the presence of increased calcium- containing stone risk |
| Age | <50 years |
Table 4: Guidelines for Surgery in Patients with Asymptomatic Primary Hyperparathyroidism [86].
a Surgery is indicated when any of these criteria is met; and also in patients for whom medical surveillance is not desired or possible
b Criteria for peri or postmenopausal women and men age 50 and older
c Premenopausal women and in men under 50, the Z-score of ≤ -2.5 is recommended as the cut-point for surgery [116] VFA-Vertebral Fracture Assessment; DEXA- Dual Energy X-Ray Absorptiometry
For those who do not meet the criteria for surgery, monitoring is recommended (Table 5). It is recommended that an annual measurement of corrected calcium, yearly or 2 yearly 3-site DEXA, and if clinically indicated, X-ray or Vertebral fracture assessment (VFA) of the spine should be performed (i.e., height loss, back pain). Creatinine clearance should be done annually and when a kidney stone is suspected, renal imaging with X-ray, ultrasound, or CT is advised. Analysis of kidney stone may be done if clinically indicated [86].
| Criteria | Monitoring |
| Skeletal | 3-Site DEXA every yearly or every other year (lumbar spine, hip regions, and distal 1/3 radius) |
| If clinically indicateda, X-ray or VFA should be done | |
| Serum | Annually |
| eGFR | eGFR annually |
| If renal stones are suspected, a 24- hour urine biochemical stone profile and renal imaging using X-ray, ultrasound, or CT scan |
Table 5: Monitoring of asymptomatic individuals with Primary Hyperparathyroidism who do not undergo surgery [86].
a Clinical indication include for example height loss and back pain; eGFREstimated Glomerular Filtration Rate; VFA-Vertebral Fracture Assessment; DEXA-Dual Energy X-Ray Absorptiometry
The rationale for monitoring is due to the fact patients may progress. Whilst 50% of patients with asymptomatic primary hyperparathyroidism will not meet any guidelines for surgery, Rubin, et al. [115] in his 15 year follow up of patients with asymptomatic hyperparathyroidism found out that many patients in the terminal 5 years of the follow up had reductions in cortical bone density and slight increases in the serum calcium concentration; and approximately 40% of patients meeting one or more guidelines indications for surgery. The only predictor of disease progression was age, where those in the average younger age group tended to progress than older age groups (52 vs 60 years old). The two studies by Rubin, et al. and Silverberg, et al. [115,117] did not reveal that mild primary hyperparathyroidism is associated with progressive renal impairment in the first 10 years of follow up; and so was the bone mineral density of the lumbar spine, hip, or distal radius. The 10-year follow study by Silverberg, et al. [117] when analysed separately, however revealed progressive disease as seen in about 25% of the cases. In particular, 4% of followed up patients developed significant worsening hypercalcaemia (>3mmol/), 15% developed marked urinary calcium excretion of >400mg/day, and about 12% exhibited decline in bone mineral density that met guidelines for surgery [117].
In the past the approach to surgery was full-neck exploration with identification and removal of the diseased gland(s). This also gave the opportunity to identify 4-gland hyperplasia or multiple adenomas, which may be found in about 15% of all cases primary hyperparathyroidism. Minimally invasive approaches are increasingly being performed due to improvements in pre-operative localization modalities and intraoperative PTH monitoring [118]. This could be done under local anaesthesia with removal of abnormal tissue without visualising the other ‘normal’ tissues. Full neck exploration surgery described earlier and minimally invasive surgeries are comparable in success rates, which is over 90% of the time [118]. Minimally invasive surgery also affords the opportunity of shorter hospital stay, more rapid recovery and in many cases it can be performed as a day case without the patient being admitted overnight. Minimally invasive techniques are also being employed in the removal of multi gland disease [119].
Common complications of parathyroidectomy include recurrent laryngeal nerve injury, hypo or hyperparathyroidism, bleeding, stridor. Intraoperative PTH level monitoring may be done, and it is prudent that PTH levels before and after gland removal is done to confirm that the adenoma removed is the true and only source of the raised PTH hormone. It is expected that within 10 minutes of the removal of the adenoma, the PTH levels should fall by at least 50%, if the adenoma is the source. If this criteria is not met, an extended exploration should be carried out to identify other possible source of the excess PTH [61,120].
When dealing with parathyroid hyperplasia such as in cases of familial hyperparathyroidism and renal failure with a four-gland disease, a sub-total parathyroidectomy with the removal of three and a half glands or a total parathyroidectomy (of all glands) followed by forearm auto transplantation of parathyroid tissue is done. The forearm provides an easily accessible site when needed. In the latter approach, it is required to have a cryopreservation facility available so that in the event of a failure of graft failure, new parathyroid tissue is available [121].
Serum calcium and PTH levels usually fall rapidly following surgery. Despite the availability of a wide array of markers, many studies have shown that bone markers decline following successful surgery. [122- 124] Markers of bone resorption also fall rapidly whilst those of bone formation decline more gradually [122], suggesting a shift in favour bone formation.
Post-operative increases in bone mineral densities have been demonstrated to occur as reported by Sankaran, et al. [125] in their meta-analysis published in 2010. Such increases have been seen at the lumbar spine, hip regions, but delayed in the distal third radius [126,127]. Increases in bone mineral density have been found to affect fracture risk as well. In a 10-year cohort study where fracture risk was evaluated, compared with those who did not undergo surgery, fracture-free survival was significantly improved with surgery [128].
The management of parathyroid cancer involves complete removal of the parathyroid cancer and local neck dissection, when indicated [52]. Neck dissection may be indicated when macroscopic characteristics of the tumour are typical of a parathyroid carcinoma, and the pathology shows extensive vascular or capsular invasion or if hypercalcaemia persists. When the diagnosis is only based on equivocal pathology in the absence of conclusive histologic features, and the patient is normocalcaemic, immediate reoperation is not indicated. In this situation regular measurement of serum calcium and PTH levels is warranted [59].
Because parathyroid carcinoma has been reported to coexist with benign adenomas or hyperplasia, all four parathyroid glands should undergo thorough exploration in patients who present with features suggestive of parathyroid carcinoma [57].
This is one of the rare cancers in which distant metastases, if present, should be considered for surgical removal [38,39]. Whilst surgery offers potential complete cure, parathyroid carcinoma has a recurrence rate of >50% [52]. Typically patients with parathyroid cancer demonstrate an indolent clinical course with disease-free intervals as long as 20 years. The management of recurrent or metastatic parathyroid carcinoma is primarily surgical [52,129,130] because chemotherapy generally is disappointing [131] whilst radiotherapy has little, if any, effect in invasive parathyroid cancer [129].
Non-surgical approaches are to be deployed among patients who do not meet the indications for surgery, refuse surgery, or when surgical and/or anesthetic risks far outweighs presumed surgical benefits [114]. This may be done by observation or employing specific medical measures to deal with complications [114]. It must be noted however that many mild diseases will progress over time [115].
It is recommended that calcium restriction should not be done. Rather, normal calcium intake should be encouraged, as in all individuals [86]. About 1000 mg/day of dietary calcium is recommended if 1,25-dihydroxy Vitamin D levels are not increased, but can be fairly restricted and tightly controlled when 1,25-dihydroxy Vitamin D is elevated [50]. This recommendation is based on the fact that urinary calcium excretion is not different in individuals with low or high calcium intake [132]; however, in the presence of elevated 1,25-dihydroxy Vitamin D, high calcium dietary intake may be associated with increased hypercalcuria [133].
It is important to maintain adequate Vitamin D levels in primary hyperparathyroidism to prevent deterioration of the parathyroid state for candidates who are being followed up without surgery [86]. Many studies have demonstrated the benefit of correcting Vitamin D deficiency and its effect on the hyperparathyroid state [134,135].
A minimum level of >20 ng/dL (50 nmol/L) of 25-Vitamin D is desirable but evidence suggests that levels >30ng/mL (75 mnmol/l) may result in further decreases in parathorme levels [86,50]. To achieve this, a starting Vitamin D dose of 800 to 1000 IU/day may help achieve this concentration [50].
Phosphate administration in the treatment of primary hyperparathyroidism is discouraged [50]. Despite the possibility of raising serum calcium levels by up to 1 mg/dL, the possible negative effects outweighs this potential benefit following long term use, including oral phosphate included limited GI tolerance, possible further increase in PTH levels, and the possibility of soft tissue calcifications [132].
Bisphosphonate therapy is the agent of choice to increase bone mineral density [86]. Among the bisphosphonates however, alendronate use has the best evidence, which improves bone mineral density at the lumbar spine without altering the serum calcium and PTH concentrations [86, 136-140].
The evidence and experience accumulated with alendronate use has made it the drug of choice in patents that do not meet any criteria for parathyroid surgery with low bone density, but not in the osteoporotic range [86].
The calcimimetic calcinet is approved drug for the medical management of hypercalcaemia associated with primary hyperparathyroidism. In many individuals, it reduces serum calcium levels to normal. It does not change bone mineral density and has modest reducing effect on parathormone levels [86,141-146]. It can also be used to treat hyppercalcamia associated with parathyroid cancers [147,148]. Currently combining bisphosphonate and cinacalcet in primary hyperparathyroidism is not recommended as there is limited data regarding its combined use [86].
Oestrogen use does not have widespread appeal in the management of primary hyperparathyroidism because of oestrogen-associated side effects [149] despite the fact that it has some beneficial use [150,151]. Though lower doses may be effective, higher intolerable doses are needed by most people to reduce serum calcium in primary hyperparathyroidism and parathormone levels do not change [152]. It also has a modest effect on the bone marrow density of lumbar spine and femoral neck of post menopausal women with mild primary hyperparathyroidism [151] making it a viable option to consider in this category of women who do not undergo surgery
Selective oestrogen receptor modulator raloxifene may be considered a useful alternate to oestrogen therapy. A study was published in 2003 [153] where 18 postmenopausal women with primary hyperparathyroidism were randomly assigned to an 8-week of 60 mg/day of raloxifene or placebo followed by a 4-week washout.
By week 8, the calcium concentration decreased significantly after raloxifene administration (10.8 ± 0.2 to 10.4 ± 0.2 mg/dl; P<0.05), markers of bone resorption and formation (osteocalcin and serum N-telopeptide) also decreased. Four weeks after raloxifene was discontinued, indices were indistinguishable from baseline. Raloxifene administration did not affect serum PTH, 1,25-dihydroxy Vitamin D, total alkaline phosphatase, or urinary calcium excretion [153]. Earlier in 2001, Zancheta, et al. [154] examined the effects of 12 months of treatment with raloxifene, 60 mg/day or 120 mg/day, on lumbar spine and femur neck bone mineral density and biochemical parameters in 3 postmenopausal osteopenic women with mild, asymptomatic primary hyperparathyroidism who had refused either parathyroid surgery or hormone replacement therapy. BMD increased by 3.4% at lumbar spine and by 2.5% at femur neck after 12 months of treatment. Total calcium and phosphate levels decreased in all the patients after 12 months of treatment. On the other hand, ionized calcium and intact parathyroid hormone levels showed a decrease after 6 months of treatment, but returned close to baseline values at month 12. It was also observed that deoxypyridinoline excretion, which is a specific marker of bone resorption, decreased by 33% after 6 months and by 61% after 12 months; fasting calcium excretion was 39% lower at 6 months and 56% lower at 12 months. Serum total alkaline phosphatase, a marker of osteoblast function, showed only a slight decrease [154].
These studies suggest that the short-term use of raloxifene may be useful in the treatment of postmenopausal women with mild primary hyperparathyroidism [153,154]; however, more long term randomized trials are needed to further elucidate its long term safety and usefulness.
Certain aspects in the diagnosis and management of primary hyperparathyroidism need further research and clarifications.
Recently, there has been the recognition of normocalcaemic primary hyperparathyroidism (NPH) and whilst it is suggested to be a variant of primary hyperparathyroidism [76,79-82], there must be further research to clarify its pathophysiology and natural history
It is unclear what truly constitutes the normal range for 25-hyroxy Vitamin D levels. This needs to be clarified to establish the level that needs to be achieved in treating Vitamin D deficiency associated with primary hyperparathyroidism to ensure optimum parathyroid levels.
We recognized that some aspects of what constitutes symptomatic and asymptomatic overlap and this may need further clarification. Monitoring for asymptomatic disease also needs to be made more definite through further research and more robust long-term data on the subject. Research focusing on cost-benefit analysis, robust long-term data and clearer recommendation in the use of non- surgical/medical modalities for the management of primary hyperparathyroidism are needed including the combined use bisphosphonate and calcimimetic (cinacalcet).
Primary hyperparathyroidism continues to be a common endocrine problem. Routine and regular calcium measurements using automated chemistry analysers over the past decades through the incidental discovery of hypercalcaemia has ensured that the nature of the presentation of primary hyperparathyroidism has been changing from bone disease and kidney stones to largely asymptomatic hypercalcaemia [61,155].
Surgery and removal of the abnormal parathyroid tissues remains the surest method of getting a permanent cure. In the past the approach to surgery was full-neck exploration with identification and removal of the diseased glands. Minimally invasive approaches are increasingly being performed due to improvements in pre-operative localisation modalities and intraoperative PTH monitoring [118]. It also has the other advantage of reduced postoperative morbidity and shorter hospital stay
Patients must be monitored for the occurrence of hypocalcaemia post-surgery and if present, must be treated with Vitamin D and calcium supplements.
The authors declare that there is no conflict of interest regarding the publication of this paper.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Yorke E, Atiase Y, Akpalu J (2018) Primary Hyperparathyroidism: an Update and In-Depth Review of the Literature. Int J Endocrinol Metab Disord 4(2): dx.doi.org/10.16966/2380-548X.150
Copyright: © 2018 Yorke E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access