Introduction
Intracranial germ cell tumors (IGCTs) represent 3-11% of paediatric
brain tumors between 0 and 19 years [1,2]. The reported incidence of
primary IGCTs in children is significantly higher in Asian countries
(especially in Japan, Taiwan and Corea) compared with Western countries
[3-5]. There is still no explanation for this geographic and ethnic difference.
IGCTs are more common in males and the peak of incidence is 10-12
years of age [1,6].
IGCTs are malignant neoplasms arising from remnants of primitive
germ cells that have failed to migrate to the genital crest during embryonic
life [7]. They show an evident predilection for the pineal region, the third
ventricle and the suprasellar-hypothalamic area, which are located
along the midline. Some IGCTs develop in unusual sites, including
the basal ganglia, thalamus, fourth ventricle, spinal cord and corpus
callosum [8-10].
Clinical presentation depends on the location and size of the tumor:
pineal region tumors can determine increased intracranial pressure due to
obstructive hydrocephalus with headache, vomit, somnolence and visual
abnormalities. Suprasellar location can cause endocrine problems due to
hypothalamic/pituitary axis dysfunction with diabetes insipidus, isolated
growth hormone deficiency, delayed sexual development or precocious
puberty and hypopituitarism. Other common symptoms include ocular
signs due to the visual pathway infiltration by the tumor [1,11-13].
Histologically IGCTs are classified in two different types: pure
germinomas, which account for approximately 50%-70% of cases, and non
germinomatous germ cell tumors (NGGCTs) which include embryonal
carcinoma, Yolk sac tumor, choriocarcinoma, teratoma and mixed germ
cell tumors. Mixed germ cell tumors, characterized by more than one
histological component, represent about 25% of all paediatric IGTCs [1].
Germ cell tumors are also classified according to tumor markers
secretion; the most common markers are β-human chorionic
gonadotropin (β-HCG) and alpha-fetoprotein (AFP), which can be
measured in both cerebrospinal fluid and serum. Pure germinomas are
usually non secreting tumors, while choriocarcinomas classically produce
β-HCG, teratomas and mixed germ cell tumors can secrete both β-HCG
and AFP [1,14].
Other several immunohistochemical markers, such as CD30, CD117,
OCT-4, β-HCG, AFP, placental alkaline phosphatase (PLAP), can be used
to determine the histological composition of the tumors [15].
Diagnosis includes radiological exams (CT and MRI), measurement
of tumor markers in blood and liquor and histological assessment [1,16].
The prognosis is closely related to the histological subtype. Pure
germinomas are highly radiosensitive, whereas NGGCTs are less sensitive
to radiotherapy and they have a worse prognosis [17,18]. The survival rate
of patients affected by pure germinoma appears to be very high, with a
10-year overall survival rate of 90%, whereas the 10-year survival rate in
NGGCTs is in the range of 30-80%, with many tumors relapsing within 18
months from diagnosis [19-22].
Also the treatment of intracranial GCTs differs according to histological
subtype. Radiotherapy, with or without chemotherapy, is the gold standard
for the treatment of pure germinomas, surgery is sometimes needed in
case of obstructive hydrocephalus [23].
Despite recent advances in chemotherapy and radiotherapy, due to its
low sensibility to this therapy, neurosurgical treatment remains essential
for the intracranial NGGCTs, providing tissue sampling, cerebrospinal
fluid diversion and cyto-reduction [24]. In particular, the recent
endoscopic endonasal transsphenoidal technique permits to approach
sellar lesions [25].
Moreover, in case of endocrine dysfunctions replacement therapies are
administrated.
We present this case in order to describe a rare form of bifocal mixed
germ cell tumor, interesting both pineal and sellar regions, with symptoms
of increased intracranial pressure, neurological disorders, endocrine
abnormalities with panhypopituitarism and a particular trend in pubertal
development that probably progressed supported by β-HCG secretion by
the tumor.
Case Report
A 16-year-old boy was referred to our Clinic with complaints of
bitemporal headache, morning vomiting, strabismus, visual disturbances
(diplopia in the lateral sight), polyuria and polydipsia since several weeks.
The clinical examination showed: weight 50.5 kg (10th centile), height
167 cm (10-25th percentile), head circumference 53.2 cm (10-25th
percentile), cardiorespiratory and abdominal examination was normal,
with no hepato-splenomegaly. Superficial lymphonodes were intact,
testicular volume was 15 ml left and 20 ml right, pubarche Tanner stage
III. Convergent strabismus due to paralysis of the left lateral rectus,
diplopia in the lateral sight, hyposthenia of the left arm, hypoesthesia on
the left hemibody was present.
Brain MRI was performed: T2 sagittal scanning showed the presence
of an expansive mass of 3 cm in diameter in the pineal region, herniating
into the third ventricle, with compression on the aqueduct of Sylvius and
secondary triventricular hydrocephalus (Figure 1); T1 weighted sagittal
scanning with gadolinium revealed that the lesion showed a remarkable
enhancement. Further lesions were present in the suprasellar region, the
genu of corpus callosum, septum pellucidum and fornix (Figure 2).
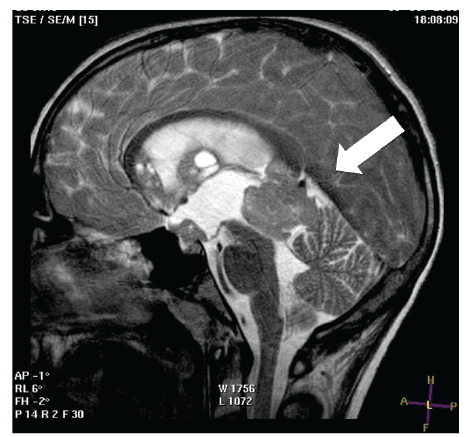
Figure 1: T1 weighted sagittal scanning with gadolinium revealed the
presence of an expansive mass of 3 cm in diameter in the pineal region,
herniating into the third ventricle, with compression on the aqueduct of
Sylvius and secondary triventricular hydrocephalus.
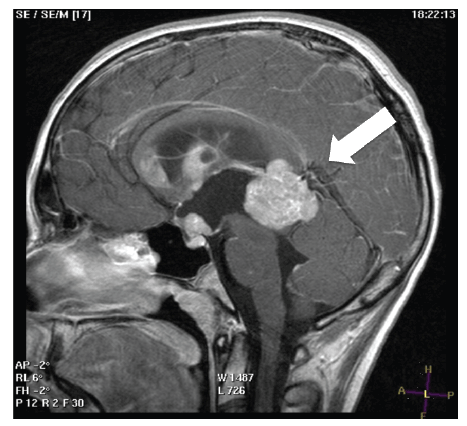
Figure 2: T1 weighted sagittal scanning with gadolinium revealed that
the lesion showed a remarkable enhancement. Further lesions were
present in the suprasellar region, the genu of corpus callosum, septum
pellucidum and fornix.
Patient was admitted and an endoscopic third ventriculostomy (ETV)
with biopsy was performed. Histological examination showed the presence
of a germ cells tumor compatible with dysgerminoma with a component
of embrional carcinoma (CD 30+ e CD 117+) (Figures 3 and 4).
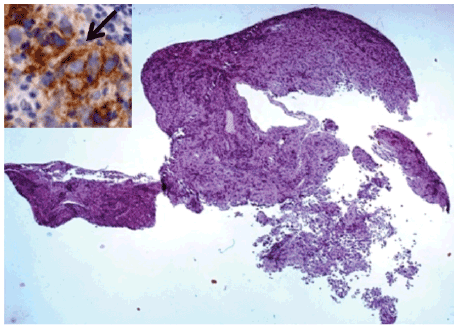
Figure 3: Mixed germ cell neoplasm at scanning power. Detail of CD117-
positive, brown stained dysgerminoma tumor cells (arrow), admixed
with embryonal carcinoma cells, which are, instead, negative for CD117,
Haematoxylin and eosin, Original magnification × 25.
Insert: Immunohistochemistry-streptavidin-biotin method; Chromogen:
diaminobenzidine; Original magnification × 200.
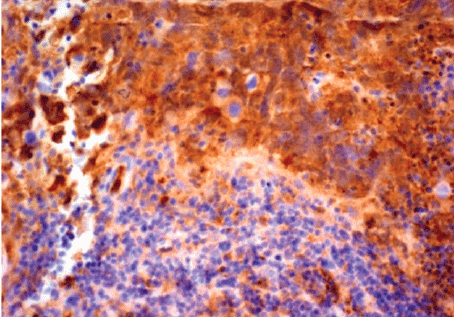
Figure 4: Component of embryonal carcinoma immunopositive for CD30.
At the bottom of the figure, dysgerminomatous cells are not labelled by
CD30 antibody.
Immunohistochemistry-streptavidin-biotin method; Chromogen:
diaminobenzidine; Original magnification × 200.
A tumor markers profile was also performed, showing elevated β-HCG
levels of 15 mUI/ml in the serum (normal value<2,6) and 19,4 mUI/ml in
the cerebrospinal fluid (normal value<15) with normal AFP levels of 2 UI/
ml in the serum (normal value<15) and 3 UI/ in the cerebrospinal fluid
(normal value<15).
Biochemistry revealed follicular stimulating hormone (FSH) 0.52
mUI/ml, luteinizing hormone (LH) 0 mUI/ml, prolactin (PRL) 33.1 ng/
ml, testosterone (T) 1030 ng/dl, estradiol 24.1 pg/ml, inhibin B<5 pg/
ml. The LHRH stimulation test did not show any increase in FSH and
LH levels. Urinary specific weight and osmolarity were extremely low.
Thyroid stimulating hormone (TSH) levels were low (0.02 µUI/mL), as
free thyroxine (FT4) levels (6.5 pg/mL). GH-IGF-1 axis investigations
revealed the presence of GH deficiency, but replacement treatments were
postponed at the end of chemo-radiotherapy.
As the patient had panhypopituitarism, glucocorticoid replacement
therapy with hydrocortisone was started (12 mg/m2
/d), followed by
levothyroxine replacement (75 µg/d), testosterone enhantate (150 mg i.m.
monthly) and desmopressin (1 puff twice daily).
After clinical-pathological-instrumental stadiation, antiblastic
treatment was started, according to the protocol of the Italian Society
of Paediatric Oncology (SIOP CNS TGC ’96). Chemotherapy was
performed for 4 cycles: cycle 1 and 3: cisplatin (changed in carboplatin
because of ear toxicity) and etoposide, cycle 2 and 4: iphosfamide and
etoposide. Chemotherapy was followed by conventional radiotherapy to
the craniospinal axis (2550 cGy), including whole ventricular irradiation
(1950 cGy) and a boost to the pineal region (900 cGy). Overall, on the
site of the primitive lesion a dose of 54 Gy was reached and at the level of
ventricles, site of metastases, the dosage was 44 Gy.
Treatment caused tumor regression, confirmed by brain MRI
performed 2 months after the end of treatment.
At the age of 17, the patient showed complete pubertal development,
weight at 50th percentile, height at 10-25th percentile.
At the last evaluation performed in our department, at the age of 18,
weight was at 25-50th percentile and height at 10-25th percentile. The
patient continued on desmopressin, hydrocortisone, L-thyroxine and GH
replacement therapies. He is currently followed at the adult’s Unit, with no
further relapses 7 years after the diagnosis.
Discussion
The germ cell tumors are rare cancers with frequent intracranial
location. The mostly affected sites are the pineal and suprasellar regions.
5%-25% of patients present synchronous lesions in both locations [1,26].
Bifocal IGCTs, by definition limited to the pineal and neurohypophyseal
regions, should not be considered as metastatic disease but as independent
primary tumors [27].
The tumors of the pineal region cause obstruction of the aqueduct
of Sylvius and foramen of Monro, resulting in hydrocephalus and signs
of intracranial hypertension; the main manifestations of suprasellar
ICGTs are endocrine diseases and visual defects. In particular, the most
common initial symptom is diabetes insipidus (50%), followed by visual
disturbances (17%). Visual defects are represented by decreased visual
acuity and visual field defects (especially bitemporal hemianopia); their
incidence is about 78% throughout the course of the disease [12].
The alterations due to hypothalamic-pituitary axis deficiency may
appear either at the onset, at overt symptomatology, during chemoradiotherapy
or during follow-up.
Our case represents a rare form of bifocal mixed germ cell tumor,
interesting both pineal and sellar regions. The patient had symptoms
of increased intracranial pressure due to hydrocephalus, neurological
disorders caused by compression by the tumor and endocrine
abnormalities with panhypopituitarism, requiring hormone replacement
therapy.
Pubertal development of our patient showed a particular trend.
Secondary sexual characteristics development was already advanced
at the time of our first observation, probably sustained by the
hypothalamic-pituitary-gonadal axis physiological activation. The
tumor situated in the pineal region has led to panhypopituitarism,
with pituitary-gonadal axis suppression confirmed by lack of FSH and
LH response to LHRH stimulation test, resulting in arrest of pubertal
development progression.
Clinically it was difficult to detect this pubertal arrest because of
secondary sexual characteristics progression, in particular pubic hair and
penile growth.
Probably this progression has been supported by β-HCG secretion by
the tumor.
The β-HCG is a molecule produced by the placenta syncytiotrophoblastic
cells; it may also be secreted in some forms of germ cell tumors. β-HCG
producing germinomas account for about 18% of all germinomas [28].
Because of its similarity with LH, β-HCG is able to bind the LH receptor
on Leydig cells and stimulate T synthesis and secretion, which inhibits LH
pituitary secretion by negative feed-back [29].
In our patient serum T level was very high at diagnosis.
Treatment caused tumor regression and suppression of abnormal
T secretion, thus stopping the progression of pubertal development,
reinstituted with replacement T therapy.
Correlation with the serum tumor markers, β-HCG and AFP is an
essential consideration in the diagnosis of IGCTs [14,30]. Detection
of β-HCG is not a marker of metastasis or tumor size, but its presence
indicates the activity of syncytiotrophoblastic elements within the tumor [31].
It is known that secreting tumors have a worse prognosis compared
to non-secretory germinomas, with a higher recurrence rate and shorter
survival [31,32].
NGGCT is diagnosed as IGCT with any malignant germ cell component
or any AFP or β-HCG secreting tumor.
The cut-off level of elevated β-HCG for the diagnosis of NGGCT differs
in each study (50–100 IU). Tumors with high β-HCG concentrations are
considered to have worse prognosis by some researchers [14,28,33,34].
However, many other studies have shown that an elevated β-HCG
level is not particularly associated with poor prognosis when there is no
evidence of dissemination through the liquor [35-39].
Conclusions
The IGCTs are very rare and require particular attention in the diagnosis
of neurological and endocrinological symptoms. The neuroimaging
characteristics of IGCTs are similar enough to limit diagnostic certainty,
therefore histological examination and measurement of specific tumor
markers are needed for a complete diagnosis [10]. Only a combined
imaging, laboratory and histological diagnosis should define the approach
that will be used [40].
One of the main challenges will be to minimize the side effects of
treatment in order to improve the quality of life of these patients.
