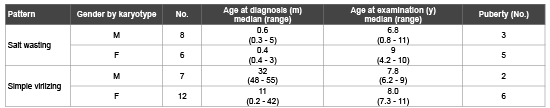
Table 1: Baseline characteristics of patients with classic 21 OH deficiency

Naglaa Fathy Barseem1* Mona F El-Samalehy2
1Pediatric Department, Faculty of Medicine, Menoufia University, Egypt*Corresponding author: Naglaa Fathy Barseem, Department of Pediatric, Faculty of Medicine, Menoufia University, Egypt, E-mail: Naglaa_b2000@yahoo.com
Background: Understanding the variation in progression from normal to precocious puberty is a matter of concern. Beside idiopathic central precocious puberty, CAH is an important cause of gonadotropin independent precocious puberty that requires a comprehensive treatment regimen to achieve normal growth and pubertal development.
Aim of the work: This study aimed to evaluate growth and pubertal changes in children with CAH. Also, to consider the idiopathic central precocious puberty.
Patients and Methods: Thirty three patients with classic 21 hydroxylase deficiency (14 SW and 19 SV CAH) enrolled in the study. They were assessed and followed up for growth and developmental characteristics .All patients were karyotyped and sex was determined, with recording of their hormonal profile and molecular genetic data, also treatment regimen was included.
Results: Mean age of pubertal onset was earlier in CAH patients; it was more evident in male patients. Sixteen of thirty three CAH patients showed signs of gonadotrophin independent Precocious puberty. At pubertal onset, mean Ht (SDS) appeared to be higher in boys especially with SV form (2.1 ± 0.2), than in girls (0.9 ± 0.1 in SW, 0.9 ± 0.03 in SV). All those children had advanced bone age at onset of puberty and within average BMI (SDS) scores. None of CAH patients had 2ry central PP except for our case report of idiopathic central precocity who required GnRH treatment. Molecular genetic study for cyp21 revealed variable sized deletions in the structure of the gene in cases with CAH, where elevated levels of 17 OH P and testosterone were additionally observed.
Conclusion: Our results demonstrated that management of CAH is challenging. Genetic studies as well as clinical, hormonal and radiological evaluation will eventually help in treatment of cases and counseling of families.
Growth; Precocious Puberty; Congenital Adrenal Hyperplasia; Cyp21; Gonadotrophin Releasing hormone
17 OHP: 17 hydroxy progesterone; ACTH: adrenocorticotrophic hormone; BMI: Body mass index; Bp: Base pair; CAH: Congenital adrenal hyperplasia; CT: Computeized tomography; cyp21: congenital adrenal hyperplasia results from steroid 21-hydroxylase cytochrome P-21; DHEA: dehydroepiandrosterone; DHT: Dihydrotestesterone; DNA: Deoxyribonucleic acid; dNTPs: Deoxynucleotide Triphosphates; F: Female; FSH: Follicular stimulating hormone; GnRH: Gonadotropin releasing hormone; Ht: Height; K: potassium; Kb:Kilo base; LH: Leutanizing hormone; M: Male; MRI: Magnetic resonance imaging; NA: Sodium; PCR: Polymerase chain reaction; PP: Precocious puberty; SV: Simple virilizing; SW: Salt wasting; Wt: Weight
Precocious puberty comprises a group of disorders that range from variants of normal development to conditions in which prompt diagnosis and therapy may be lifesaving. The line separating normal from abnormal onset of puberty represents children entering puberty more than 2.5 standard deviation earlier than average [1-3]. Precocious puberty can be classified as whether it is true or central; that is due to early but otherwise normal activation of hypothalamic pituitary gonadal axis, or pseudo precocious puberty i.e. for other causes due to production of gonadal steroids and if it is of the same or contra lateral sex [4]. An important cause of precocity in boys and girls is congenital adrenal hyperplasia (CAH) that refers to an autosomal recessive condition caused by enzymatic deficiency involving the steroidogenesis pathway [5]. The most common form of which is 21 hydroxylase deficiency; caused by mutations in cyp21 gene [6], this gene encoding human 21 (OH) (cyp21) is located at 6 p 21.3 within the HLA class III, of ten exons, approximately 30 Kb apart from its inactive pseudogene; (cyp21 p) with different mutations within its structure and then malfunction [7]. The clinical phenotype is determined by the degree of enzymatic activity that includes the more severe salt wasting (SW) form, the less severe simple virilizing (SV) and the mild non- classic one [6]. Pubertal development seems to occur along a continuum from normal to precocious which implies the importance of etiological evaluation, together with close monitoring and follow-up of these children with either true or pseudo precocity and great direction towards counseling with their families [8].
The aim of this work was to evaluate the pubertal changes in children with classical congenital adrenal hyperplasia, also, to take in consideration the importance of idiopathic central precocity.
The present study was conducted on thirty three patients with CAH. They were selected from Genetics and Endocrinology unit, pediatric department, Menoufia university hospitals and Menoufia medical Insurance hospital, Egypt. They were fifteen males and eighteen females (14 SW and 19 SV). Their ages ranged from one week to eleven years. All patients with classic 21 OH deficiency were being followed in our unit during the period from 2008 till near the beginning of 2016. Patients’ demographic data, family pedigree construction, evaluation of growth and pubertal characteristics together with hormonal and biochemical profile were done, also included regimen of treatment, after obtaining a written consent from patients’ families. Data were documented using report sheets. The studied patients were subjected to the following:
For anthropometric measurements, weight and height were individually measured at different ages. Body mass index (BMI SDS) was calculated from weight (kg) divided by height (m2 ) (Kg/m2 ) and expressed as SDS [10].
- Phallus: size, length, degree of differentiation, and location of the urethral meatus.
- The scrotum or labia for: fusion or not of two halves of labioscrotal tissue or pigmentation.
- Gonadal exam: with stress on their site; within or outside the labioscrotal tissue, their size and texture in apparent male cases.
- Prader score application for degree of virilization in apparent female cases [11].
- DNA extraction: genomic DNA was extracted from peripheral whole blood by DNA purification kit that uses a convenient spin column technology according to manufacture protocol (thermo scientific GeneJET Tm kit, # k0721 made in EU. Lithuania).
- Polymerase chain reaction: selective PCR amplification of the CYP 21 gene in two segments; where segment 1 represents a 1339-bp extended from exon 1 to exon 6 and segment 2 is a 2220-bp extended from 8–bp deletion in exon 3 to exon 10 according to Oriole et al. [16].
Each PCR reaction was performed with mixtures of 0,2 mg of genomic DNA, 2.5 µ of Taq DNA polymerase, 0.5 mml/ l of each forward and reverse primer, 100 mmol/l dNTPs, 3.0 mm MgCl2 and 1x PCR buffer to a final volume of 50 ml reaction volume. PCR cycling conditions were 5 min at 94.c for initial denaturation, followed by 35 cycles at 94.c for 1 min, 65.c for 1 min, and 72.c for 6 min, plus cycle elongation of 2 s for each cycle, and 72.c for 15 min for final extension. PCR was performed using an automated thermal cycler (Bio- Rad MJ Research PTC-200 DNA Engine thermal cycler).
- Primer sequence used for amplification of the two segments of the studied gene were [16]:
For segment (1):
p1 (5- TCGGTGGGAGGGTACCTGAAGGT- 3)
p2 (5– GCATCTCCACGATGTGAT- 3).
For segment (2):
p3 (5– CCGGACCTGTCCTTGGGAGACTACT- 3)
P4 (5– CTGAGCGGCTGGGTGAAATGGAAC -3)
- The PCR reaction products were electrophoresed in an automated ABI prism sequencer (applied biosystems). For amplification of a corresponding exon, allele specific primers were used as previously described [17]. Results were analyzed with free software gene marker 1.6 (Soft Genetics, State College, PA, USA).
Patients with classic 21 OH Deficiency were enrolled and followed up. They included (15 boys, 18 girls). Fourteen patients had SW-CAH, and nineteen patients had SV-CAH. Sixteen patients had entered puberty, two cases reached final height. The age at presentation ranged from weeks up to eleven years. Consanguinity was evident in eleven cases, while FH of CAH was observed in nine cases. Three cases had other siblings affected with CAH. These baseline characteristics were shown in table 1. Tables 2 and 3 showed clinical and anthropometric parameters of patients with classic 21 OH deficiency ,in addition to changes regarding age at puberty, bone age advancement and need for GnRH treatment for cases who entered puberty, while, biochemical hormonal profile, imaging and results of molecular genetic studies were shown in table 4. External genital manifestations in female patients showed variable degrees of virilization (prader score 1-3) was mainly clitoromegaly with fused labia and no palpable gonads. For male patients, external genitalia showed skin pigmentation, normal male external genitalia with variable length of penis and palpable gonads. All patients had elevated levels of 17 OH P and testosterone during their pubertal period. Results of genetic study were shown in table 3. Pelviabdominal imaging studies revealed internal female genitalia in all genetic females; on the other hand, all genetic males had no internal female genital organs. Normal MRI findings were present in the case report of idiopathic central precocity. For growth at onset of puberty, mean Ht (SDS) was higher in SV boys (SV 2.1 ± 0.2, SW 1.1 ± 0.04) than girls (SV 0.9 ± 0.03, SW 0.9 ± 0.1). BMI (SDS) was within average in both sexes of CAH. Tanner stage (PH2) occurred at mean age of; (in boys SV 5.8 ± 0.1, SW 6.8 ± 0.1), and (SV 7.1 ± 0.3, SW 8.6 + -0.02 in girls) respectively, and earlier age of onset of puberty.
Table 1: Baseline characteristics of patients with classic 21 OH deficiency
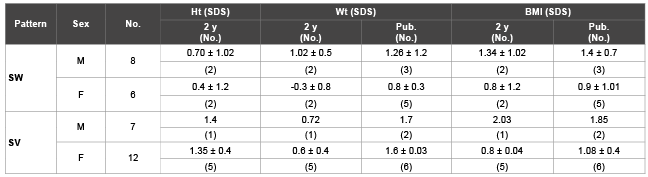
Table 2: Clinical data of patients with classic 21 OH deficiency
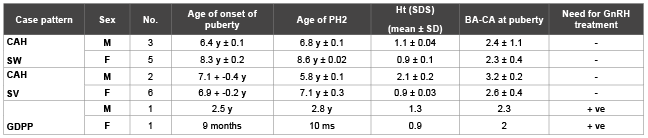
Table 3: Pubertal changes in patients with precocious puberty
BA-CA: Bone age–Chronological age.
GnRH: Gonadotrophin releasing hormone
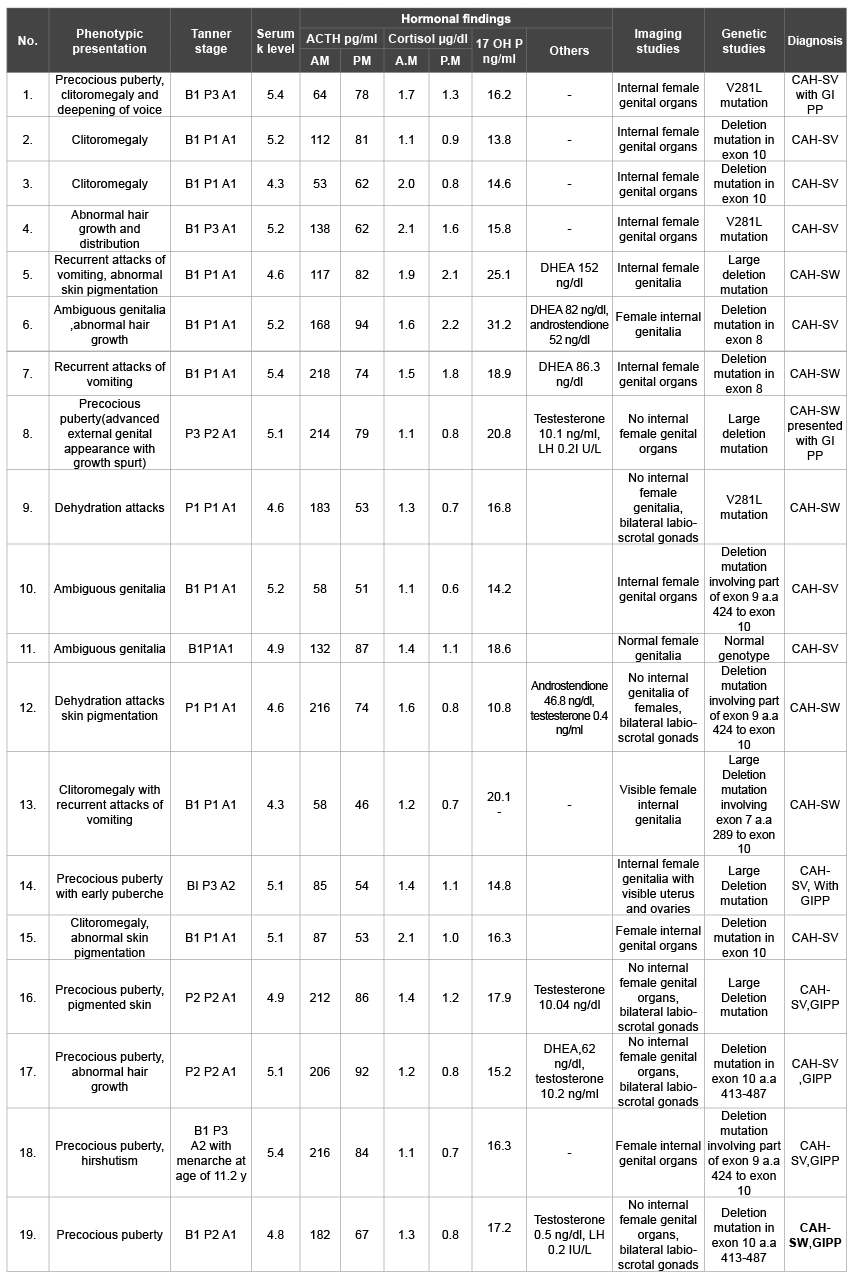
Table 4: Summarized data of patients underwent molecular study
CAH: Congenital adrenal hyperplasia
GIPP: gonadotrophin independent precocious puberty
a.a: amino acid
SW: Salt Wasting
SV: Simple virilizing
Added to these cases of CAH, one case report of gonadotrophindependent precocious puberty where; 9 month - aged female patient, the second to a mother aged 26 yrs and father of 36 yrs. The first sibling is 3 year-aged normal boy with irrelevant family history of similar conditions, complained of mother’s notification of vaginal bleeding which started at the age of 6 months with no history of trauma. According to mother’s statement, there was normal vaginal delivery, no history of incubation, and average birth weight. No minor or major anomalies were present. Regarding the vaginal bleeding, it stayed for about 4 days, and recurred again. This finding was preceded by appearance of pubic and scanty axillary hair but it did not make sense to the mother until the occurrence of vaginal bleeding, for which the mother sought medical advice and the patient was investigated. On examination, she had unremarkable neurological findings, non palpable thyroid gland, absent skin pigmentation. Examination of the breast revealed enlarged both breasts with firm consistency and developed nipple and areola, no discharge (Figures 1 and 2). As regard anthropometric measurements; she weighed 10 kg (0.8 SDS), her length was 75 cm (2 SD for age), corresponding to 0.9 SDS. She had tanner stage of A1 P2 B3 with two attacks of vaginal bleeding. Pelvic abdominal ultra sound showed adult sized uterus with pubertal size, shape and proportions, thin endometrial central line and uterine length of 43 mms (enlarged for age; i.e length >34 mms). In this scan, both ovaries are pubertal with bilateral (almost adult sized) follicular pattern, no related masses or cysts. Skeletal maturation was assessed by x-ray on Lt wrist, using Greulich and pyle [13]; the bone age of the patient was at 2 years (Figure 3) which is greater for her chronological age. MRI of the brain was done and no abnormalities were detected. As for hormonal assay, serum estradiol and LH, FSH levels were elevated and within the pubertal range; E2 was 48.0 pg/ml (ref. range 6.0-27.0), serum LH and FSH levels during GnRH stimulation test were 76.4 mIu/ml and 67.8 mIu/ ml 4h post decapeptyl respectively.
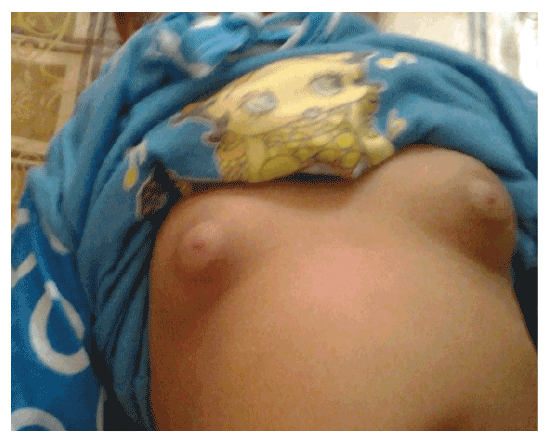
Figure 1: Photographs at 9 months of age showed breast development consistent with the degree of secondary sexual maturation.

Figure 2: Photographs at 9 months of age showed pubic hair development consistent with the degree of secondary sexual maturation.
All investigations came to a belief that this patient had idiopathic central precocious puberty. After counseling with her parents we started treatment in the form of GnRH agonist; Sc injection of decapeptyl 3.6 mg every 4 weeks to be continued for 11 years of her age and be followed up. Unless the patient started her medical treatment, she would be waiting for her third monthly interval of menstrual cycle. This case report emphasized how this condition should be evaluated through clinical, biological and radiological examination and properly managed.
Controversy is present about the normal age of onset of puberty in boys and girls [18-20]. In this study, the pubertal onset was significantly earlier in patients with both forms of CAH, compared with the observed normal reference [21]. Regarding growth in patients with CAH, ten patients aged less than 2 years; their Ht (SDS) was comparable to normal reference pattern in all groups of patients. This was not in accordance with Balsamo et al. [22]; who showed that in both sexes, the average length of patients with classic CAH was greater than the mean birth length in controls. He stated also, among CAH patients, those with SW form was longer but weighed less than those with SV form. On the other hand, in our study it was found that, at pubertal onset, mean Ht (SDS) appeared to be higher in boys especially those with SV form. Final height was reached in two female SV cases that were in the lower range of normal Egyptian females. This finding showed agreement with a multicenter analysis study which showed that pubertal growth is more impaired in patients with SV –CAH compared to SW- CAH patients [23].
About genital examination, Tanner stage revealed prepubertal testicular size of 3.5-4 ml, pubic hair stage 3 with slightly enlarged phallus in three pubertal male cases, plus breast stage 2 in pubertal girls with CAH. This is consistent with the findings of Emans and Jean [24] that clarified the hormonal effect on secondary sexual characters. Menarche occurred at the age of 11, 2 in two cases with SV form, which was comparable to reference age among Egyptian girls [21]. Most of our patients had advanced bone age at onset of puberty that might reflect the integrated effect of dose and duration of sex steroid action during pre-pubertal period which associated with poor height outcome. This is in accordance with Mogonsen et al. [25]; who stated that CAH may result in rapid growth and precocious puberty with premature skeletal maturation. We found that none of our CAH patients had true precocious puberty so, did not require treatment with GnRH analogue; a consequence of excess of adrenal androgens, leading to early activation of the hypothalamic pituitary gonadal axis and occurrence of secondary central precocious puberty [26,27]. All patients in our study received hydrocortisone tablets according to our unit protocol and underwent follow- up measurement of 17 OHP and testosterone levels for treatment monitoring. These two steroids notably 17 OHP appeared to be markedly elevated during puberty; this is the basis of successful treatment of CAH which depends on achieving the delicate balance of suppressing adrenal androgen secretion while maintaining normal growth [27-29]. Family history was relevant in nine cases, so it is recommended that three- generational family pedigree and consanguinity should be obtained and structured. All of these are constant with the fact that CAH is autosomal recessive [30]. This emphasizes the importance of CAH screening programs together with molecular genetic testing especially when future pregnancies are anticipated.
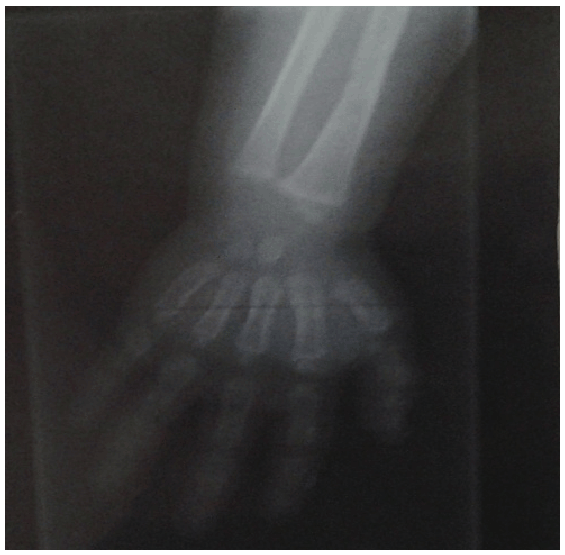
Figure 3: Photograph showed osseous maturation, advanced for her chronological age.
The prevalence of different forms of CAH as well as their mutation pattern varies among ethnic populations Example of which are studies in Iran, Turkey, East India and Tunisia, where the latter showed high frequency of large deletions and Q318X mutations in Tunisian population [17].
As regard the molecular study for cyp 21 gene, variable sized deletions, observed in our patients, were the most relevant mutations. Substitution mutations (V 281 L) were found in three cases, despite its association for non – classic forms of CAH [31]. One case had normal genotype; 8-bp deletion mutation was not detected in our patients. These mutations in the gene caused alterations in the function which ultimately affected enzyme activity of 21 OH, of which its deficiency is the commonest cause of CAH [32,33]. As in our case reports, several reports showed that although the rarity of the constitutional type of central precocity, it should be kept in mind [34].
Precocious puberty is such a serious problem that girls and boys who began puberty long before their peers may be extremely self conscious about the changes occurring in their bodies. This may affect self- esteem and increase the risk of depression, so therapeutic approach remains to be determined [35].
Continuous clinical practice shows variations in the expression of precocious puberty in children. Results of this study focused on the importance of monitoring of growth parameters and bone age as determinants of Precocious puberty, resulted from inadequate hormonal control of CAH with both clinical; SW and SV forms. These findings clarified that early diagnosis towards establishing effective screening program is crucial.
Download Provisional PDF Here
Article Type: Research Article
Citation: Barseem NF, El-Samalehy MF (2016) Pattern of Pubertal Changes in Congenital Adrenal Hyperplasia Patients, with Idiopathic Central Precocious Puberty; Case Report. Int J Endocrinol Metab Disord 2(1): doi http://dx.doi.org/10.16966/2380-548X.121
Copyright: © 2016 Barseem NF, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access