Introduction
Steroidogenic factor-1 (SF1/AD4BP/FTZF1) plays a key role in the
regulation of adrenal and reproductive organs differentiation. The SF1
gene (NR5A1) is an autosomal gene located on chromosome 9q33 and it is
the member 1 of the nuclear receptor subfamily 5, group A [1]. It expands
over 30 kb of genomic DNA and it is comprised of one non-coding exon
(I) followed by six coding exons (II to VII).
The protein consists of 461 amino acids that include a DNA-binding
domain (DBD) containing two zinc fingers, a hinge region containing
a first functional activation domain (AF-1), a ligand-binding domain
(LBD) of 12 helices (H1-H12) that includes a second functional
activation domain (AF-2), and an accessory region [2]. It is expressed
in undifferentiated gonads even before SRY and it is necessary for testis
determination and differentiation. SF1 protein is highly expressed in
steroidogenic tissues, such as gonads, adrenals, and placenta, and regulates
almost all the enzymes related to this process. It is also expressed in the
ventromedial hypothalamic nucleus and pituitary gonadotropes with
relevant physiological roles in the central nervous system [3].
Homozygous null mice (Nr5a1−/−) have adrenal and gonadal agenesis,
persistent Müllerian structures in XY karyotype, partial hypogonadotropic
hypogonadism, and other features such as hyposplenism, abnormalities of
the ventromedial hypothalamus, and late-onset obesity [4,5].
In humans, mutations in NR5A1 were reported to cause both 46,XY and
46,XX gonadal dysgenesis with or without adrenal failure [6-8]. Variable
loss of SF1 function is associated with a wide phenotypic spectrum
indicating that SF1 dosage is critical. In the approximately 81 patients so
far described with impairment to the protein function mutations in the
NR5A1 gene the clinical phenotype is largely variable including, female
genitalia, mild clitoromegaly, isolated hypospadias, anorchia and even
normal male genitalia with infertility [6,9]. In 46,XX affected female
patients, mutations in NR5A1 are associated with different phenotypes
including primary and secondary amenorrhea, premature menopause
and decreased ovarian reserve, but preserved fertility [7,8,10].
We are reporting the clinical phenotype, hormonal pattern and
molecular studies (genetic, functional data and protein structural
analysis) in two non-related 46,XY, DSD index patients and their affected
family members. A novel heterozygous NR5A1 gene mutation, c.909G>A
(p.Ser303Arg), and a previously described [8,10] heterozygous NR5A1
gene mutation, c.938G>A (p.Arg313His), both located in exon 5, in the
highly conserved helix 5 (H5) of the ligand binding domain of the protein
(LBD), are presented. Functional studies and protein structure analyses
shed light into the reasons sustaining the pathogenicity of the mutations.
Subjects and Methods
Clinical material
Two families with an index case presenting with a 46,XY DSD were
evaluated. Four generations of Family 1 and two generations of Family 2
are depicted in the family tree of Figure 1.
The clinical and biochemical findings in the affected members of the
first family kindred have been previously reported [10]. Family 2 has not
been previously reported.
This study was approved by the Ethics Committee of the Garrahan
Pediatric Hospital. Written informed consent for the study was obtained
from all adult patients or patients´ parents or tutors.
Mutation analysis of the NR5A1 gene
Genomic DNA was extracted from peripheral blood leukocytes by
standard procedures. The coding exons (exons 2-7) and flanking intronic
regions of the NR5A1 gene were amplified by PCR using specific primers
gently provided by Dr. J. C. Achermann, University College London, UK.
The PCR products were purified (Qia Quick PCR purification kit, Qiagen,
Buenos Aires, Argentina) and sequenced using a BigDye Terminator
version 3.1 cycle sequencing kit (Applied Biosystems, Buenos Aires,
Argentina) on an ABI PRISM 3130 Genetic Analyzer capillary DNA
Sequencer (Applied Biosystems, Buenos Aires, Argentina). The primers
used for sequencing were the same as those used for PCR. The nucleotide
sequences obtained were compared with the NCBI entry of NR5A1 gene:
NG_008176.1.
In silico protein analysis
The sequence homology-based tool SIFT (Sorting Intolerant from
Tolerant; http://sift.jcvi.org/), version 2.0.6 and the structure-based
tool PolyPhen-2 (Polymorphism Phenotyping v2; http://genetics.bwh.
harvard.edu/pph2/) were used to predict the pathogenicity of the missense
variants p.Arg313His and p.Ser303Arg using default settings. The original
sequence of the protein was obtained from the Ensembl and Uniprot/
Swiss-Prot databases.
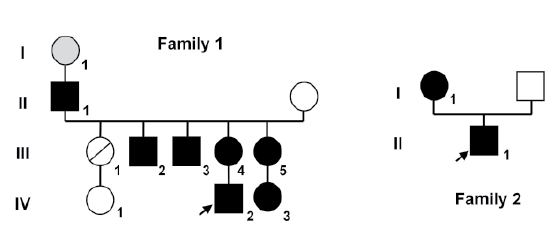
Figure 1: Family trees of both kindreds. Squares represent males and circles represent females. Black squares represent affected 46, XY subjects
who were raised as boys, and black circles represent affected 46, XX subjects. Index case in each family is indicated by an arrow. Family 1 the circle
with a slash through it represents a deceased female who was not studied. Subject I-1 (gray circle) experienced menopause at the age of 30 years
and was inferred to be affected (not available for study).
Site-directed mutagenesis
For promoter activity experiments, an expression vector containing
human wild type SF1 cDNA (p-SF1wt) was constructed in the
pcDNA3 vector (Invitrogen, Buenos Aires, Argentina). The SF1
cDNA was PCR amplified using specific primers, SF1for_pcDNA3
(5′TGGTGTGAGGGGGTTTCTG3′) and SF1rev_pcDNA3
(5′GAGAGGAGGAAGGGATGACC3′) carrying EcoRI and Xho1
restriction enzyme sites, from RT-PCR of H295R cell line (human
adrenocortical carcinoma cell line). The 1921-bp PCR product was 2%
agarose gel-purified using the Zymoclean Gel DNA Recovery Kit (ZYMO
RESEARCH, Buenos Aires, Argentina). The fragment was digested with
EcoRI/Xho1 and cloned into the EcoRI and Xho1 sites of the pcDNA3
vector. The accuracy of the construct was confirmed by sequencing.
NR5A1 expression vectors containing the p.Arg313His and p.Ser303Arg
variants were generated by PCR-based site-directed mutagenesis
(QuikChange Site-Directed Mutagenesis Kit, Stratagene, Buenos Aires,
Argentina) with specific primers, using p-SF1wt as a template (p-R313H
and p-S303R). The entire sequence of all mutant plasmids was confirmed
by direct sequencing prior to functional studies.
In vitro functional studies of NR5A1 mutations
All transient gene expression studies to assess NR5A1/SF1 function
were performed in 24-well plates using Lipofectamine 2000 reagent
(Invitrogen. Buenos Aires, Argentina) according to the manufacturer’s
protocol. Each expression vector (p-SF1wt, p-R313H or p-S303R) was
co-transfected into SMAT1 cell line (murine immature Sertoli cells)
or into Y1 cell line (murine adrenocortical tumor cells) with reporter
plasmid (PGL2, Promega, Buenos Aires, Argentina) containing hAMH
promoter kindly provided by Dr. Rodolfo Rey (Centro de Investigaciones
Endocrinológicas (CEDIE), Hospital de Niños R. Gutiérrez, Buenos Aires,
Argentina), p-PhAMH or with reporter plasmid (PGL3, Promega, Buenos
Aires, Argentina) containing the h3BHSD2 promoter, p-Ph3BHSD2. Both
promoters have response elements for SF1. The cells were lysed 48 hrs after
transfection and assayed for luciferase activity (Dual Luciferase Reporter
Assay System; Promega, Buenos Aires, Argentina). Co-transfection of
CMV Renilla luciferase was used as a marker of transfection efficiency.
Results are shown as the mean ± SEM of three independent experiments,
each performed in triplicate and represents the ratio of luciferase activity
of each SF1 and mutants compare to the empty vector. All data were
standardized for Renilla activity. Statistical significance was examined
using ANOVA test. Values of p<0.05 were considered significant.
In silico structural analysis of the wt protein and variants
Protein complexes in solution were prepared using as scaffold the
X-ray crystallographic structure 1ZDT (chain B) of human SF1 wt LBD
bound to a co-regulator peptide (NCoA-2, KENALLRYLLDKD) and
a phospholipid ligand (di-palmitoyl-3-SN-phosphatidylethanolamine,
PEF). Ser303Arg and Arg313His point mutations were introduced in
silico with Discovery Studio Visualizer 3.5 [11]. Protonation states of
the ionizable residues at pH 7.4 were defined by visual inspection after
addition of H atoms missing in the 2.10 Å resolution crystal structure.
Each structure was solvated by a truncated octahedral box of TIP3P
water extended up to 12 Å around each complex, adding K+ ions to
electroneutrality (7, 6, or 8 counterions respectively for wt, p.Ser303Arg
and p.Arg313His). A standard classical minimization protocol (2500 steps
relaxing solvent plus ions while maintaining the complex restrained with
500 kcal/mol.Å, followed by 20000 steps of unrestrained optimization of
the whole system) was applied under periodical boundary conditions with
the sander module of the AMBER12 suite [12], assigning the ff03 force
field to both SF1 and the co-regulator peptide, and the GAFF parameters
to the phospholipid ligand. RESP atomic charges were also obtained
with Gaussian09 rev. A.02 [13]. Long-range electrostatics was treated
using the Particle-mesh Ewald (PME) approach [14], and a 10 Å cut-of
was used for direct space interactions. Molecular dynamics simulations
(MD) were run in explicit water for each complex at 300 K under the
same conditions, using a 2 fs integration step and applying restrictions
to H atoms with SHAKE algorithm [15]. After 100 ps equilibration in a
NVT ensemble using a Langevin thermostate, 5 ns of NPT simulations
were produced with pmemd from the AMBER12 suite of programs [12].
MD trajectory post-processing was conducted with the ptraj module of
AmberTools 12 [12] and representative structures corresponding to the
most populated cluster (among five clusters generated with the averagedlinkage
algorithm) were obtained for each complex.
Results
Clinical features
The index case Family 1 [10], patient IV-2 was born with ambiguous
genitalia (2 cm length phallus, labioscrotal folds, severe hypospadias, and
small inguinal gonads). Hormonal determinations revealed elevated serum
FSH and low AMH levels, and normal steroidogenic response to hCG
stimulation (Table 1). Male sex was assigned. Laparoscopic examination
and bilateral orchidopexy was performed at 10 months of age. Müllerian
structures were observed and a biopsy of the right gonad revealed signs of
testicular dysgenesis, absence of Leydig cells and atypical heterochromatic
germ cells. No lesions of carcinoma in situ were observed. Three other
46,XY individuals in the family (subjects II-1, III-2 and III-3) presented
with severe hypospadias at birth, all of them developed spontaneous male
puberty, and one (subject II-1) has fathered 5 children. Among the family
members one 46,XX individual (subject I-1) developed early menopause
(this subject was not available for the biochemical and genetic evaluation)
and other three 46,XX family members (subjects III-4, III-5 and IV-3)
presented high FSH and low AMH levels.
The index case of Family 2 (patient II-1) was the first child of nonconsanguineous
parents. The baby was born with ambiguous genitalia
and was initially assigned the female sex at birth. The patient was referred
for further evaluation at three months of age. On physical examination,
the baby presented a 2.5 cm length phallus with well developed corporal
tissue, complete labioscrotal fusion and scrotal hypospadias; both inguinal
gonads were palpable. Hormonal determinations revealed elevated serum
FSH and low AMH levels, as well as normal steroidogenic response to hCG
stimulation (Table 1). Sex was re-assigned to male. Müllerian structures
were present on ultrasound and confirmed by laparoscopy. A right gonad
biopsy at 10 months of age revealed mild testicular dysgenesis, presence of
Leydig cells, and few germ cells. His mother reported difficulties getting
pregnant and presented low AMH and high basal FSH serum levels.
His father referred normal sexual development and fertility. All affected
individuals studied presented normal adrenal function.
Mutational analysis
The NR5A1 gene molecular study revealed the mutation c.938G>A
(p.Arg313His) in heterozygous state in the first family and a novel
mutation, c.909G>A (p.Ser303Arg) in heterozygous state in the second
family. Both mutations are located in exon 5 in the highly conserved H5 of
the LBD. In order to determine if these alterations are present with a high
frequency in the general population (Single Nucleotide Polymorphism
Database), we looked for these variations in the NCBI databases and
ensembl genome browser and we did not find them, suggesting that these
two variations would not be common polymorphisms. In addition, we
search for the mutations in 100 healthy subjects (200 alleles) by DNA
sequencing and no allele carrying this mutation was detected.
NR5A1 gene mutation prediction model
To assess the potential deleterious effect of the amino acid changes,
we used two software programs, SIFT and PolyPhen-2. SIFT prediction
is based on the degree of conservation of amino acid residues in sequence
alignments derived from closely related sequences. Arginine residue in
position 313 and serine residue in position 303 are highly conserved
between species. These mutations were evaluated to the option “affects
protein function” with a highly deleterious tolerance index score of 0.00 and
0.02 respectively. The same assessment was performed with PolyPhen-2.
This software predicts the possible impact of an amino acid substitution
on the structure and function of a human protein using straightforward
physical and comparative considerations. Both mutations were predicted
to be as “probably damaging” with a score of 1.000 and 0.999 respectively.
Structural analysis
The two variants containing point mutations p.Ser303Arg and
p.Arg313His were compared to human SF1 wt. Both amino acid changes
are located in the H5 helix of the LBD. In particular, p.Ser303Arg is
located in the vicinity of the area of interaction with the co-regulator
peptide (mainly delimited by H12, H3, and H4) and the p.Arg313His
mutation is part of a salt bridge H2…H5 compacting the LBD structure of
the receptor and stabilizing its active form [2,16].
From a comparison of the dynamical behavior of the protein backbone
at the three SF1 complexes in solution emerges that whereas p.Ser303Arg
displays a similar structural evolution than that of SF1 wt, for the
p.Arg313His variant it takes longer (near 4 nanoseconds (ns)) to stabilize
the 3D structure, which globally differs from the other two variants due to
the loss of the salt bridge interaction (Figure 2A).
Since it has been reported that restrictions in the flexibility of NR5A1
receptors knock down their activation [17,18], we have compared this
issue between the three variants using the root mean square fluctuations
of the protein by residue (RMSF) calculated at the stabilized part of each 5
ns simulation (Figure 2B). As it can be immediately appreciated, whereas
the p.Ser303Arg mutation turns the protein significantly more flexible
throughout all its structure as compared to the wt (particularly in the
H2-H3 loop and in the H12 helix), the p.Arg313His mutation turns, in
contrast, the receptor more rigid in several regions.
The overall comparison of the tridimensional arrangement of alphahelices
and loops of the LBD across the three variants (Figures 2C and
2D) highlights that both point mutations induce changes in the tertiary
structure of the receptor around the ligand (H2-H3/H6-H7 loops, beta
hairpin, and H6/H7 helices) and in the AF-2 domain (H4, H11, and H12),
resulting the latter in a different positioning of the co-activator peptide.
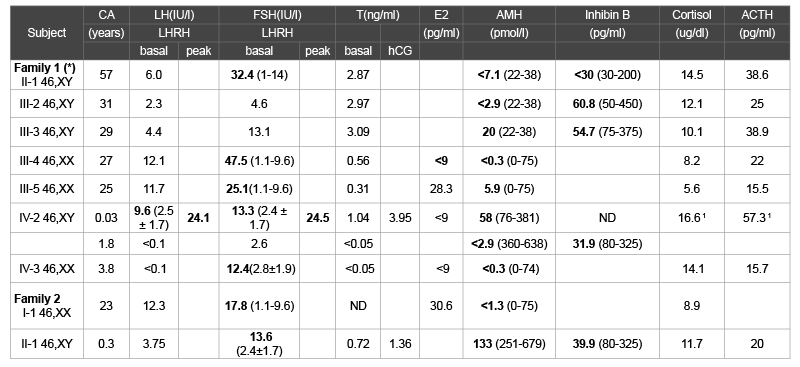
Table 1: Serum hormone levels in the affected individuals available for the study.
Boldface numbers indicate values outside the reference range; normal values are shown in parentheses. SI conversion factors: testosterone (nanomoles
per
liter), 3.47; estradiol (picomoles per liter), 3.671; cortisol (nanomoles per liter), 27.59.
CA= Chronological age. ND=Not determined
1at the age of 0.83 years
(*) Ciaccio et al. [10]
A closer inspection of the hydrogen-bond network surrounding each
mutation promptly shows that whereas the interaction with Lys434 is the
most affected for p.Ser303Arg (in comparison to SF1 wt), Arg313 ability of
establishing hydrogen bonds with Glu237 and Asp309 in wt is completely
lost when replaced by His313 in p.Arg313His (Table 2).
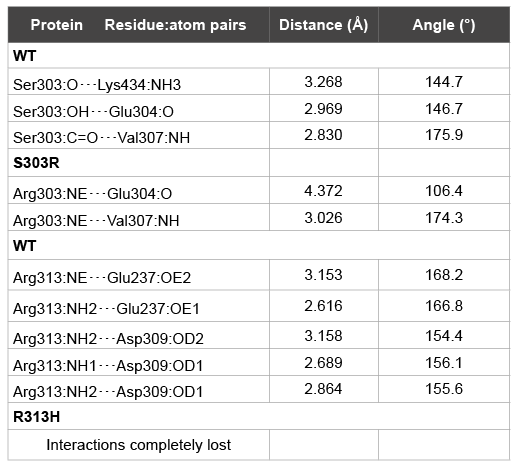
Table 2: Structural analysis of the hydrogen bond interactions around each
point mutation
Finally, a closer look in the comparison of the interactions with the
ligand at the LBD across variants shows that as the p.Ser303Arg mutation
affects SF1 interaction with the phospholipid mainly through changes in
Lys440 positioning and the opening of the channel’s entrance defined by
H2-H3 and H6-H7 loops (this resulting in PEF’s polar head more exposed
to the solvent), p.Arg313His mutation does not produce significant
changes on the ligand-receptor recognition (Figures 2E and 2F).
Functional studies of NR5A1 mutation activities
To examine the transcriptional activity of the SF1 mutants (p-R313H
and p-S303R) and the wild type SF1 (p-SF1wt), we performed transient cotransfection
assays with different SF1 target gene promoters (p-PhAMH
and p-Ph3BHSD2) in steroidogenic cell lines (SMAT1 and Y1).
In both steroidogenic cell lines, p-SF1wt significantly (p<0.05)
increased reporter gene expression driven by the human AMH or
3BHSD2 promoters. Transfection with each of the two missense mutations
p-R313H or p-S303R showed impaired transactivation activity compared
with p-SF1wt (Figure 3).
In details, in the SMAT1 cell line the co-transfection with p-PhAMH
and p-SF1wt significantly increased the luciferase activity (3.00 ± 0.09 AU,
mean ± SEM, p <0.05 ANOVA) while mutants p-R313H and p-S303R
significantly decreased luciferase activity compared to p-SF1wt (1.32
AU ± 0.05 and 1.09 AU ± 0.01 respectively, mean ± SEM, p<0.05
ANOVA) (Figure 3A). Similar results were obtained in the SMAT1 cell
line with the co-transfection with p-Ph3BHSD2; a significant increased
luciferase activity in the presence of p-SF1wt (6.92 AU ± 0.25, mean ±
SEM, p<0.05 ANOVA) while when assaying the effect of the mutants
p-R313H and p-S303R a significant reduction of transactivation
capacity was observed compared to p-SF1wt (1.91 AU ± 0.07 and 2.00
AU ± 0.08 respectively, mean ± SEM, p<0.05 ANOVA) (Figure 3B).
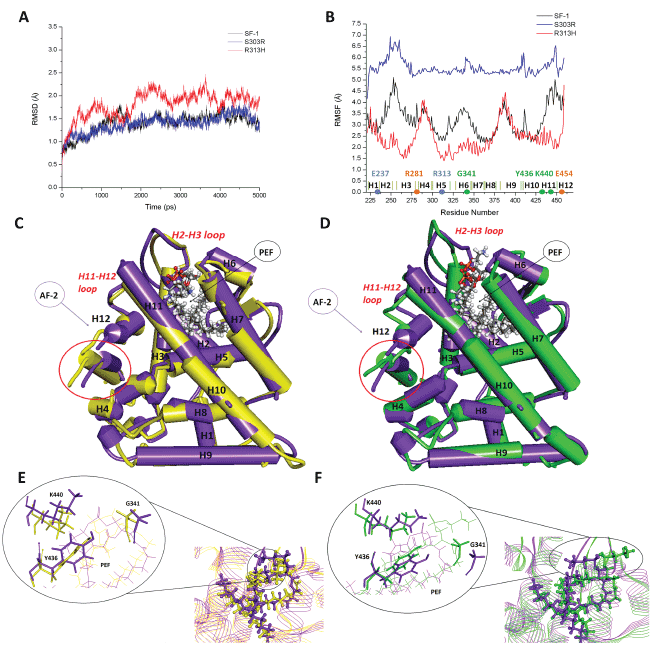
Figure 2: Structural analysis of SF1 mutations. Panel A - Structural evolution of Cα RMSD for each SF1 variant along the simulation taking the
initial structure as reference. Panel B - Flexibility by protein residue calculated as RMS fluctuations from the time-averaged Cα positions along the
stabilized part of each simulation. Panels C and D - Overlay by pairs of protein variants, evidencing the main differences on α-helices, β-hairpin,
and loops 3D disposition in the LBD. In purple the SF1 wt protein, in yellow the R313H mutant, and in green the S303R mutant. AF-2 is the liganddependent
activation function and PEF a phospholipid ligand. Circled in red is the coactivator peptide. Panels E and F – Detail of the region around
the ligand, compared by pair of protein variants. In purple the SF1 wt protein, in yellow the R313H mutant, and in green the S303R mutant.
In the Y1 cell line, similar responses as in the SMAT1 cell line were
observed with p-Ph3BHSD2 and p-SF1wt (9.27 AU ± 0.54, mean ± SEM,
p<0.05 ANOVA) and mutants p-R313H and p-S303R (4.55 AU ± 0.32 and
4.08 AU ± 0.17 respectively, mean ± SEM, p<0.05 vs. SF1wt, ANOVA) (Figure
3C). Transactivating studies of both mutants with the p-PhAMH in the Y1
cell line were not possible as the p-SF1wt did not increase luciferase activity.
Discussion
We are reporting the molecular characterization of two missense
mutations (p.Arg313His and p.Ser303Arg) in the LBD of the SF1 gene, in
two non-related 46,XY DSD patients, both in heterozygous state.
The clinical and biochemical phenotypes of these two 46,XY index
patients were similar. Hormonal determinations showed normal basal and
peak testosterone, high serum FSH and low serum AMH levels for the
child’s age. Evidence of testicular dysgenesis and the presence of Müllerian
structures suggested a failure of embryonic fetal Sertoli cells to secrete
AMH during the sensitive prenatal period, which is consistent with the
low levels of AMH detected during infancy. The baby of the second family,
evaluated at the age of minipuberty, also presented low serum inhibin
B levels along with elevated FSH. As it was proposed in an aromatase
deficient boy, inhibin B might be the major contributor in the regulation
of serum FSH secretion in normal infant males [19]. Both patients were
initially assigned the female sex at birth, but after a careful evaluation, male
sex re-assignation was recommended to, and accepted by the parents, on
the basis of adequate basal T secretion for age, including normal response
to hCG stimulation, and potential for successful intercourse in adulthood.
This decision was also supported by the follow-up of other 46,XY DSD
members of the first family who developed spontaneous male puberty,
one with preserved fertility [10] and lack of reports of development of
testicular tumors in 46,XY NR5A1 dysgenetic testes.
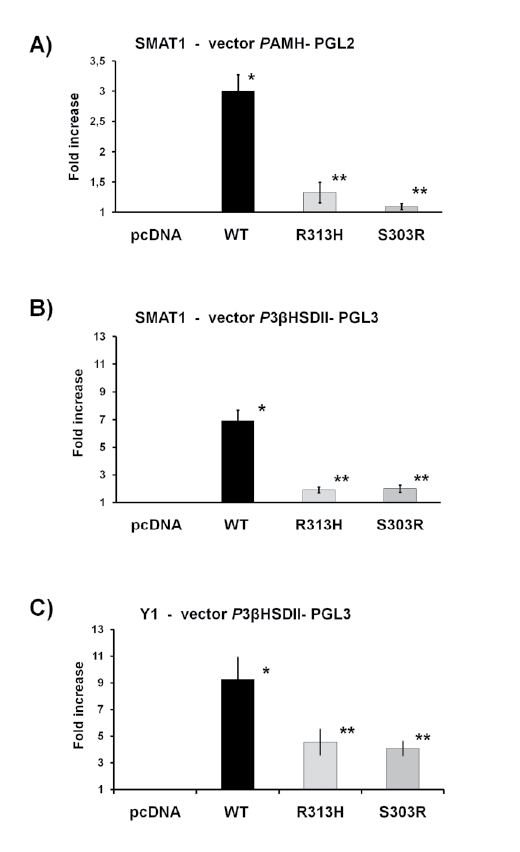
Figure 3: Functional analysis in SMAT1 and Y1 of the R313H
and S303R mutations. The transcriptional activity of wild-type SF1
(p-SF1wt) and mutants p.Arg313His and p.Ser303Arg (p-R313H and
p-S303R respectively) were studied using Ph3BHSD2 and PhAMH
promoters in SMAT1 cell line and Ph3BHSD2 promoter in Y1 cell line.
Both mutants in both cell line and with both promoters exhibited a
reduction of transactivation activity. The results are expressed as fold
increase of luciferase activity compared with empty vector (mean ± SD).
All values represent the means ± SEM of three separate transfection
experiments, each performed in triplicate. * vs. empty vector; ** vs. SF1,
p<0.05, ANOVA.
There was a striking variability among the affected relatives in the
first family that range from severe ambiguous genitalia to normal male
external genitalia and preserved fertility, as it was previously reported by
us [10]. A wide phenotypic spectrum has been described in patients with
heterozygous NR5A1 mutations including when the mutation is located
in the LBD (Table 3) [5,6,9,20-32]. Phenotype variability in NR5A1
mutations makes genotype-phenotype correlations very difficult. In 46,XX
individuals carrying heterozygous NR5A1 mutations, variable gonadal
phenotypes have also been described [7,8,10,20,33-36]. In this report,
four 46,XX affected women presented high serum FSH levels with low
levels of AMH. The presence of regular menses in the three young-adult
women studied and successful maternity in two, reflect a compensated
ovarian dysfunction. These women may go through a stage of decreased
ovarian reserve before developing clinical signs or symptoms of ovarian
failure, as it has been reported [20].
Adrenal function was normal in both index patients and their relatives
carrying mutations in NR5A1. To date, adrenal insufficiency was reported
in only three patients harboring NR5A1 mutations [37-39].
The amino acid substitution of both missense mutations (p.Arg313His
and p.Ser303Arg) takes place in a highly conserved site among species
observed by in silico analysis.
The structural analysis showed that, since it turns to be a less rigid
protein, interactions are expected to be more labile for p.Ser303Arg
mutant. On the other hand, the loss of flexibility induced by p.Arg313His
mutation occurs in regions which are involved in several interactions
of relevance for structural packing (H2-E238 and H5-R313) and coactivator
recruiting at the activation function domain AF-2 (H3-R281
and H12-E454). The H11 C-term region where the phospholipid polar
head binds at the entrance of the ligand channel is also highly affected. In
agreement with this, crystallographic studies on the SF1 ligand binding
domain revealed different phospholipids bound in its hormone binding
pocket [2,16-18,40-42].
In vitro assays used to assess the functional impact of these two
mutations, on h3BHSD2 and hAMH promoters and in two different
steroidogenic cell lines (SMAT1 and Y1), showed impaired transactivation
activity. On this line, besides the known interaction with phospholipids,
its LBD might interact with some unknown co-activators to trigger
adrenal and gonadal development [43]. Even though the in vitro studies
in some reports of NR5A1 gene mutations located in the LBD, confirm
the deleterious effect (Table 3), the clinical phenotype variability observed
among the affected patients reported remains poorly understood.
Even though, SF1 is known to be engaged in the interaction
with numerous co-activators acting over the promoter region of
several steroidogenic enzymes and factors involved in reproduction,
steroidogenesis and sexual differentiation [1,6,39], the molecular
mechanisms of heterozygous mutations remain to be elucidated.
The majority of the patients previously described, including the
present report, carried heterozygous NR5A1 gene mutations in the LBD,
supporting the concept of dose dependence of SF1 action (Table 3).
In summary, we are reporting, in two non-related 46,XY DSD patients,
the clinical phenotype, hormonal studies, molecular characterization,
and protein structural analysis of one missense mutation of the NR5A1
gene (p.Arg313His) previously described without any functional study
performed, and a novel missense mutation (p.Ser303Arg), both in
heterozygous state, located in the LBD. In vitro and in silico experiments
argued for their functional impact and also provided insight into the
structure-function relationship of the SF1 protein. Finally the present
study reinforces the concept of the wide variability in the clinical
phenotype in affected 46,XY DSD patients.
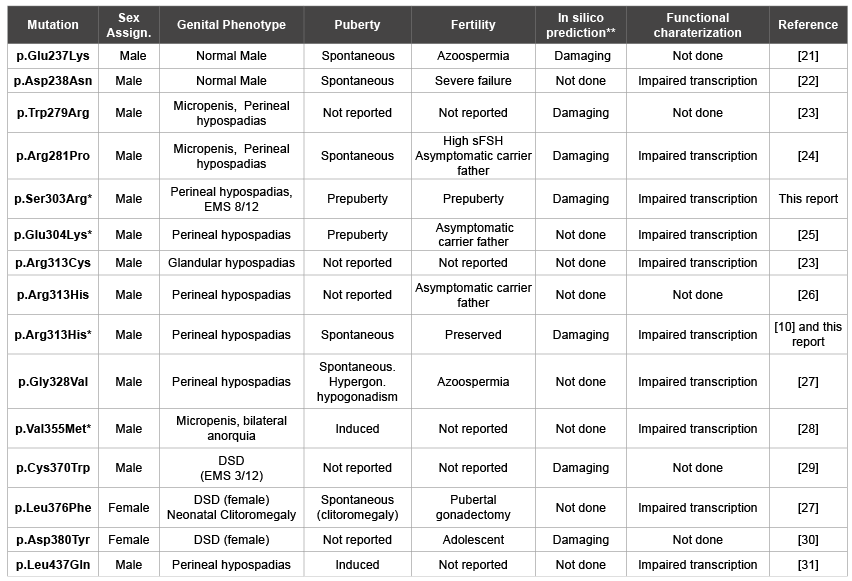
Table 3:Missense mutations in the SF-1 ligand binding domain of 46,XY patients
*In silico structural analysis performed;
**Analyzed using SIFT and/or PolyPhen-2 tools.
sFSH: serum FSH.
EMS: External Masculinization Score (minimum: 1, maximum: 12) [32]
Acknowledgement
Supported by grants from Consejo Nacional de Investigaciones
Científicas y Técnicas (CONICET), Fondo para la Investigación Científica
y Tecnologica (FONCYT), Argentina.
