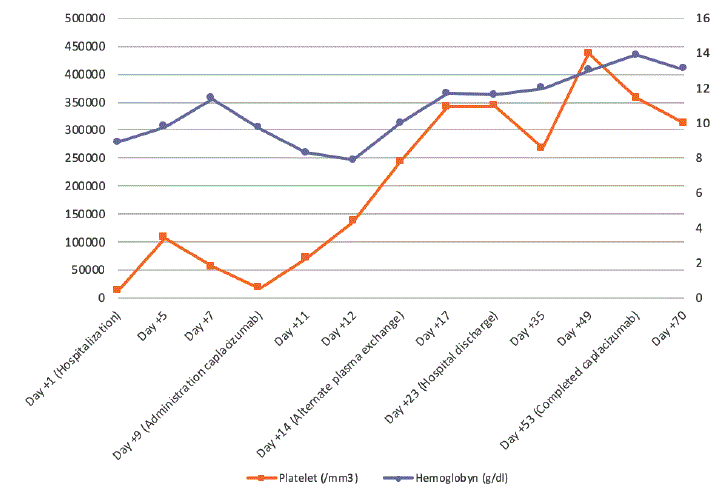
Figure 1: Platelet and hemoglobyn levels.

Lara Martín Rizo1* Noemí Gómez García1 Laura Llorente González2 Alfons Serrano Maestro2 Miriam Sánchez Mateo1 Jaime Pérez de Oteyza2 José Manuel Ortega Gómez1
1Pharmacy Department, Hospital University HM Sanchinarro, Madrid, Spain*Corresponding author: Lara Martín Rizo, Pharmacy Department, Hospital University HM Sanchinarro, Street Oña 10 Madrid, Spain, E-mail: lara. martinrizo@gmail.com
Thrombotic thrombocytopenic purpura is a thrombotic microangiopathy, caused by a deficiency of ADAMTS-13 (a desintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). It is considered a medical emergency and its standard treatment is based on plasma exchange associated with high doses of corticosteroids. Those patients refractory to standard treatment have a high risk of morbidity-mortality. Through this clinical case, a patient refractory to all the treatments is presented. She was treated whit caplacizumab that is humanized bivalent antibody acting against the domain A1 of the von Willebrand factor and prevents interaction with the glycoprotein Ib-IX-V (GPIb-IX-X). After one year from the end of treatment, the patient remains stable and asymptomatic. Caplacizumab is a good choice in those patients who do not respond to any of the standard treatments and that can lead to a decrease in mortality in patients with refractory thrombotic thrombocytopenic purpura.
Caplacizumab; Refractory; Thrombotic thrombocytopenic purpura; Antibody; Platelets
Thrombotic Thrombocytopenic Purpura (TTP) is a thrombotic microangiopathy, caused by a severe deficiency of ADAMTS-13 (a desintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) which leads to platelet consumption in von Willebrand factor-platelet aggregates and microvascular thrombosis [1].
Under normal conditions the von Willebrand Factor (VWF) acts as a central protein of primary hemostasis. In the event of vascular injury, VWF makes the union between the damaged endothelium and the GPIb-IX-X. The endothelium secretes the ultra-long multimeric von Willebrand factor (VWF-UL) in response to damage, and later they are fragmented by protein ADAMTS-13 [2].
ADAMTS-13 deficiency can be either congenital or acquired. Congenital TTP is caused by a constitutional deficiency of ADAMTS-13, while idiopathic TTP is due to the auto antibodyinhibited function of ADAMTS-13 [3]. The organism produces antibodies that attack ADAMTS-13; this causes an increase in the VWF-UL which has an affinity for platelets and give rise to aggregates that cause thromboembolic complications [4].
This pathology is considered a medical emergency with a mortality of 80-100% without proper treatment [5]. It is characterized by: microangiopathic anemia, thrombocytopenia, fever, neurological involvement, renal insufficiency and deficit of ADAMTS-13 below 10%. The incidence is 4.5 per million people and year [6]. Testing for ADAMTS-13 activity and autoantibodies directed against ADAMTS-13 is standard to diagnose TTP [7].
There are guides focused on the diagnostic study and treatment of this pathology but there is no specific guide available for urgent treatment during the first hours of your suspicion [8]. There is a certain consensus where it is determined that the standard treatment should be based on plasma exchange [4]. In fact, the plasma exchange began to be used for PTT in 1970s based on observations that blood or plasma transfusion for anemia increased the platelet count of the patients [9]. Plasma infusion or exchange therapy increased the survival rate from less than 10% to approximately 60% and 80%, respectively [10].
Daily plasma replacement should be performed until response is obtained. According to the 2012 American Society of Apheresis Consensus Conference on TTP, an increase in platelets two consecutive days greater than 150.000, normal Lactate Dehydrogenase levels (LDH) and improvement of neurological symptomatology, is considered response [11].
A plasma exchange is associated with corticosteroids (prednisone 1mg/kg or dexamethasone 40 mg/day for 4 days) as an immunosuppressant to avoid the production of antibodies [12].
The efficiency of plasma exchange in removing ADAMTS-13 antibodies varies because it is affected not only by diffusion from their extravascular pool but also by newly synthesized antibody molecules from plasma cells. Consequently, the time to normalized platelet count ranges broadly from a few days to more than a month [13].
Also, approximately 10% to 20% of TTP patients show lack of response [14]. Refractoriness to standard treatment is described as a platelet fall after 4-7 days of plasma exchange or deterioration in the clinical status. In this case, the number of plasma exchanges can be increased or resort to more powerful immunosuppressants such as rituximab, cyclophosphamide or vincristine [11].
The experience with humanized and bivalent antibody caplacizumab is presented in a patient with TTP refractory to all therapies marketed so far.
38-year-old woman went to the emergency department for asthenia and mild headache of weeks of evolution. In the last 48 hours she began with petechiae and non-traumatic hematomas in the body. She presented with choluria but there was no history of dysuria or pain. There was no history suggesting the involvement of respiratory, cardiovascular and gastrointestinal symptoms.
At admission she presented hemoglobin (Hb) of 8.9 g/dl, platelets of 13000/mm3 and lactate dehydrogenase (LDH) of 1593 U/L. The peripheral blood smear had 4.2% schistocytes and a direct negative antiglobulin test.
Admitted for suspected TTP due to microangiopathic hemolytic anemia (observed through the smear), thrombocytopenia and high LDH. The patient began treatment with corticosteroids 1 mg/kg/ day and daily plasma exchange by extending the initial study with hormones, tumor markers, rheumatic tests, serology, immunology, urine analysis and ADAMTS-13 determinations. The value of the latter was less than 1%.
On day +7 post-diagnosis worsening was observed with platelet fall, maintained Hb and persistence of schistocytes. Treatment resistance was detected without clinical repercussion. Corticosteroids were increased and rituximab was associated weekly (375mg/m2) and vincristine 2 mg. In addition, treatment with acetylcysteine 300mg/ day was started as an off-label use. The patient received two doses of rituximab in total: One on day +7 and one on day +14.
By not getting a response, maintaining very low platelet levels and observing schistocytes in the peripheral blood smear, it was decided to administer a single bolus of cyclophosphamide 800 mg/m2.
On day +8 there was no response; the medical team contacted the pharmacy service to request caplacizumab.
Caplacizumab is a humanized and bivalent antibody of the variable domain of the immunoglobulin that acts against the domain A1 of the VWF and prevents interaction with GPIb-IX-X. Its action prevents VWF-UL from forming aggregates with platelets that are the cause of microthrombic production [1].
After conducting a literature search, it was found that the drug was in phase III and that it could be available through compassionate use. The pharmacy service reached the laboratory which indicated the procedures to follow. The laboratory requested a series of data regarding the admission and follow-up of the patient (ADAMTS-13 determination, platelet count at admission or current symptomatology). After approval by the hospital’s medical management, the drug was ordered very urgently through the medication page in special situations of the Spanish drug agency.
After 24 hours the medication arrived at the pharmacy service and on the day +9 post-diagnosis the administration of caplacizumab began.
Caplacizumab administration was performed following the indications provided by the laboratory: the first dose of 10 mg was administered intravenously before performing the first plasma exchange of the day. From the next day and maintaining the 24-h periodicity, 10 mg were administered subcutaneously at the end of all plasma changes performed on the day. In principle, the forecast was to administer the drug up to 30 days after the last plasmapheresis.
The patient experienced a clear improvement 48-72 hours after the beginning of the administration, which was observed in the progressive increase in the number of platelets, as can be seen in figure 1.
Figure 1: Platelet and hemoglobyn levels.
Due to this clear improvement, the pattern of corticosteroids was decreasing in successive days maintaining daily plasma replacement and the weekly rituximab. On day +14 plasma exchanges is scheduled on alternate days maintaining clinical improvement and progressive increase of platelet and Hb count until stabilization in normal values as seen in figure 1.
On day +23 the last plasmapheresis was performed; the corticosteroid was suspended and the patient was discharged. Caplacizumab treatment was maintained at home, to help the patient with the administration; it was trained by the pharmacy service together with the nursing team of the day hospital for the correct drug administration at home. An educational talk was held on the correct handling, conservation and administration of the drug.
Caplacizumab was maintained daily until 30 days after the last plasmapheresis as scheduled (day +53 post-diagnosis). Weekly appointments were scheduled from the pharmacy service, coinciding with the appointments in the hematology service, to dispense the medication and monitor the evolution.
During home follow-up, the patient has remained stable and asymptomatic. Only experienced heavy bleeding coinciding with menstruation that produced a decrease in hemoglobin without transfusion. Currently the patient remains asymptomatic and she continues with her periodic reviews with the hematology service. Table 1 summarizes the most important aspects of the clinical case.
| Day of ingress | Evolution |
| Day+1 | Suspected TTP. Treatment corticosteroids and plasma exchange |
| Day+3 | Good treatment response. Progressive increase in the number of platelets |
| Day+6 | Decreased number of platelets. Maintenance of Hb levels and persistence of schistocytes |
| Day+7 | Treatment resistance |
| Rituximab 375 mg/m2 | |
| Vincristine 2 mg | |
| Acetylcysteine 300 mg/day | |
| Day+8 | Cyclophosphamide 800 mg/m2 |
| Request Caplacizumab | |
| Day+9 | Caplacizumab administrations |
| Day+10 | Caplacizumab administrations after last plasma exchange of the day |
| Day+11 | Symptomatology improvement Progressive increase in the number of platelets |
| Day+14 | Alternate plasma exchange |
| Second dose of rituximab 375 mg/m2 | |
| Day+17 | Stabilization of platelet level |
| Day +23 | Last plasma exchange |
| Corticosteroids suspension | |
| Hospital discharge | |
| Day +35 | Clinical improvement after discharge |
| Day+53 | Completed caplacizumab |
Table 1: Most important aspect of patient treatment.
This clinical case attempts to show the benefit that humanized antibodies against the A1 domain of VWF can exert in patients refractory to standard therapies that have a significant risk of complications that even lead to death.
The plasma replacement manages to eliminate the ultra-long VWF and replace ADAMTS13 and corticosteroids inhibit the formation of antibodies against ADAMTS13. However, they are slow-acting treatments that do not prevent platelet aggregation and they maintain the formation of microthrombi being able to cause the death of the patient [2]. Also, the immunosuppressive treatments are effective only for a small fraction of patients.
Caplacizumab may be an alternative in those patients with refractory TTP, always associated with standard treatment. Further investigation is needed to determine whether caplacizumab in monotherapy or in association with other drug, can meet the ultimate goal of eliminating the need for plasma therapy [13].
Access to the drug through compassionate use has allowed the treatment of this patient, being the first patient treated in Spain outside of a clinical trial. Phase III has shown that caplacizumab prevents platelet aggregation more quickly compared to standard monotherapy treatment. This helps prevent final ischemic injury to the organs in the short and long term [15].
It has also been seen that it normalizes the platelet count in a shorter period of time than those patients receiving only standard therapy since caplacizumab prevents platelet consumption and prevents the development of refractory disease [1].
Tasai HM, in the study, concluded that with precautions, caplacizumab together with rituximab is expected to reduce the burden of exacerbation, refractory or persistent disease, recurrence, and possibly death [13].
In our patient a clear improvement is observed from the beginning of caplacizumab, reaching good platelet values around the day +12 postdiagnosis (day +3 from the beginning of caplacizumab). It is true that the patient had received several treatments, among them rituximab, in a short period of time. The association between caplacizumab and rituximab could be responsible for the increase in platelet levels in our patient.
In the phase II clinical trial, the persistence of ADAMTS13 deficit was seen below 10% in a subgroup of patients after 30 days of treatment with caplacizumab. Therefore, during the development of phase III, the option of continuing treatment with caplacizumab was established after 30 days if there was a persistence of autoimmune activity [1].
In our case the patient did not require caplacizumab after day +30, maintaining stable platelet levels and absence of signs of disease.
Regarding adverse effects, caplacizumab treatment presents risk of bleeding: Epistaxis and gingival bleeding occurred in 23 and 13 patients, respectively, in the caplacizumab group in fase III. Our patient showed good tolerance to treatment, although she had heavy bleeding coinciding with menstruation that could be related to treatment with caplacizumab.
After one year from the end of treatment, the patient remains stable and asymptomatic, with no signs of relapse and normal platelet levels.
It can be concluded that caplacizumab is an alternative therapeutic in those patients who do not respond to any of the standard treatments. In our case, the patient recovery was temporally associated with the administration of caplacizumab. It is essential to understand the medical urgency of this diagnosis and the importance of a fast and coordinated work between hematology and hospital pharmacy services to achieve good results quickly and effectively for the benefit of the patient.
Conception and design: L.M.R., N.G.G.; Analysis and interpretation: L.M.R., N.G.G., L.L.G; Drafting and critical revision of the article: L.M.R., N.G.G., L.L.G, A.S.M., M.S.M., J.P.O., J.M.O.G.; Final approval of the article: L.M.R., N.G.G., L.L.G, A.S.M., M.S.M., J.P.O., and J.M.O.G.
This research was funded by Sanofi Aventis S.A.
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Rizo LM, García NG, González LL, Maestro AS, Mateo MS, et al. (2020) Caplacizumab: First Case of Thrombotic Thrombocytopenic Purpura Refractory Treated in Spain outside Clinical Trial. J Drug Res Dev 6(2): dx.doi.org/10.16966/2470-1009.153
Copyright: © 2020 Rizo LM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access