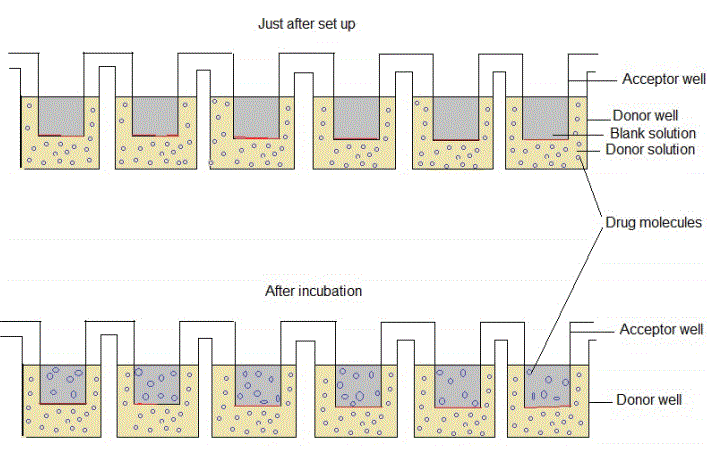
Figure 1: The PAMPA set up before and after incubation.

Andrew G Mtewa1,2* Kennedy Ngwira3 Fanuel Lampiao4 Anke Weisheit2 Cassim U Tolo2 Patrick E Ogwang2 Duncan C Sesaazi2
1Department of Applied Sciences, Malawi University of Science and Technology, Malawi*Corresponding author: Andrew G Mtewa, Department of Applied Sciences, Malawi University of Science and Technology, Malawi, E-mail: amtewa@must.ac.mw; andrewmtewa@yahoo.com
Potential drug molecular leads from plants are usually tested for their efficacy in bioactivity assays in preclinical trials. This is followed by testing for the properties of the molecular structures, if they can be bio-available to biological targets, should they proceed to clinical trials. Most drug molecules with proven bioactivities fail to qualify as potential drug candidates due to their poor molecular drug-like properties. It is therefore imperative for drug developers and discoverers to start focusing on the molecular structural properties at an early stage to decide whether the particular drug molecule, with bioactivity, is worthy investing on. This review aims at putting together and discussing fundamental methods for selected drug-like properties; permeability, pKa, LogP and LogDx . It was noted that basic methods on the aforementioned procedures are being customized for simplicity and convenience, mainly in the form of commercial in silico innovations. There is a need for cheaper methods to be developed to ease budgetary constraints on drug discovery and designing. This report will provide pointers to choose appropriate methods in drug designing and development, making the whole process more convenient and relatively cheaper for researchers, students and research funding bodies. Due to technical variations that each method has, reporting of results on these properties should be reported along with specific methods and conditions used.
Permeability; pKa, LogP and LogDx; Drug development; Drug designing
The selection of appropriate drug molecule candidates and their optimization from plants are some of the most critical stages of drug discovery and development. Traditionally, bioactivity studies alone used to be recognized as the major factor in the selection of drug molecule targets, but recently, it is accepted that this is not enough without understanding the ‘drug-like’ properties that the molecules portray [1]. Two complementary approaches are now available to drug discoverers; molecular bioactivity and molecular property-based approaches. To narrow down to a good choice of drug-like candidates, property-based approach provides a guide towards suitable and successful bioactivity strategies [2,3]. Bioactivity based approaches in the preclinical stages make use of in vitro and in vivo studies, though some [4] suggest a reduction in the reliance of in vitro studies as they do not represent the actual physiological environment but only delays the drug discovery process. Drug-like properties are properties of those molecules that have sufficiently and highly acceptable Absorption, Distribution, Metabolism, Elimination and Toxicity (ADME/Tox) properties to proceed through to the completion of clinical trials, phase I [5]. They are recognized to have major effects on drug performance on biological targets [6]. Their studies help in the optimization of Pharmacokinetic (PK) aspects of drugs, thereby increasing the success rates of the drug molecules under development [7].
Drug-like properties, such as solubility, permeability, metabolic stability and transporter effects are of critical importance for the success of drug candidates mainly in the early stages of drug discovery [8]. They affect oral bioavailability, metabolism, clearance, toxicity, as well as in vitro pharmacology. Bioavailability refers to a measure of the extent and rate at which an active drug molecule takes to reach a biological target for action [9].
It should be noted that insoluble and impermeable compounds can result in erroneous biological data and unreliable Structural Activity Relationships (SARs) in enzyme and cell-based assays and also rapid metabolism by enzymes and high efflux by transporters can lead to high clearance, short half-life, low systemic exposure and inadequate efficacy [6]. It is reported that most of the drugs from preclinical tests fail to successfully make it through the entire clinical trials phase due to poor molecular drug like properties including permeability, lipophilicity and pKa[10]. Only 8% of new drug chemical entities that reach phase I of clinical trials for Central Nervous System (CNS) for example, successfully get clinical approvals [10]. Early property information helps teams make informed decisions and avoid wasting precious resources on candidates that will likely not pass clinical trial phase I. Structure-property relationships are essential to guide structural modification to improve properties. High throughput ADME/TOX assays have been implemented and are being widely used to drive drug discovery projects in parallel with activity screening [6]. It is therefore necessary to put together knowledge of fundamental methods of drug like properties’ determination and outline their advantages and disadvantages so to provide options for researchers, students and research administrators in the best choices of methods at their disposal, making research and research decisions easier and more efficient and convenient.
Drug pKa is a measure of the extent to which a particular drug molecule will ionize in various pH environments and has a direct influence on many Pharmacokinetic (PK) parameters [11]. The following methods describe key features in the methods for its determination.
Capillary electrophoresis for pKa determination: This is a high throughput method in the measurement of pKa which relies on the electrophoretic mobility differences in the retention time of a compound in its ionized and neutral forms [12]. An aqueous buffer is used as a solvent to dilute the test compound. The test compound is then run repeatedly using capillary electrophoresis mobile phase buffers. The mobile phase buffers are prepared at different pH values. Since the capillary electrophoretic mobility is directly proportional to charge, molecules that are ionized are made to move faster through the mobile phase. The effective mobility is monitored through retention time and it becomes shorter progressively with the increase in ionized molecule fractions. A plot of effective mobility against the pH of the mobile phase leads to the determination of pKa , which is at the point of inflation [6]. The advantage with this method is that it has very low impurity interferences due to its high separation capabilities [13] but the whole setup is usually hard to come by for financially challenged laboratories.
Spectral Gradient Analysis (SGA) for pKa determination: This method takes a chromatographic approach. The concept of operation is similar to the one in gradient high pressure liquid chromatography pump, only having the two liquid phases replaced by aqueous basic and acidic buffers. Dimethylsulfoxide (DMSO; 10 mM) is used to dissolve the test compound and the solution is placed in a 96-well plate. Each of the solution is diluted with the buffers mentioned above. A program is then run on the test sample solutions to ensure gradient mixing from one buffer at a higher percentage to the lowest within 2 minutes, and then goes to the next buffer. This makes pH to be changing continuously, consequently, changing the fraction of the compound being ionized. Visible or UV chromophore absorption near the ionization center (in the distance of within 3 to 4 bonds) gets to change with ionization [14]. Thereafter, the UV-Vis absorption changes with pH as the solution mixture moves into a diode array UV detector. The inflation point of this absorption curve is the pKa of the test compound [15,16]. This method requires a high capital investment but provides high throughput as well, with pKa screens being produced every 3 to 4 minutes [6]. It is also to be a reliable method with results comparable to other methods [14].
Potentiometry in pKa determination: In this method, water is usually used as a solvent for the test sample. The dissolved test compound is titrated with a basic or an acidic buffer with a known concentration (the titrant). The setup involves dipping a pH meter electrode in the test compound solution to monitor pH progression. As the titrant is added to the solution with test compound, the pH of the solution gets to change accordingly. A plot of pH change with titrating equivalents against pH of the solution leads to the determination of pKa , which is essentially the pH at the point of inflation of the plotted curve from the Henderson-hasselbalch equation [17,18]. Ionization extent can alternatively be determined by measuring the UV absorbance at a point close to the ionization center. Potentiometric method in the determination of pKa is considered a gold standard due to its flexibility [19].
Permeability is a measure of how much a drug molecule penetrates through membranes of various phases and is key to drug distribution and absorption. Most permeability tests are done for oral drugs [20] with a few for topical administration. The determination of permeability is more complex than most drug-like properties, which is the reason for limited number of measurements available for algorithm development [21]. Permeability methods usually provide different results from laboratory to laboratory due to variations in validation protocols [6]. It is therefore always necessary that whenever inter laboratory tests are being conducted for verification purposes, the methods and conditions should be identical. In silico, in vivo and in vitro methods are available for use. In vivo methods usually have higher reliability as they present ideal drug environments required for the target. However, due to high costs, slower speed and labor demanding nature of in vivo measurements, their application in the selection of drug leads molecules and their optimization proves to be very challenging [22]. The other challenge with in vivo measurements particularly using rodent models is that permeability results are usually over estimated as drug compounds’ penetration rates are higher potentially due to possible permeation through hair follicles [23]. In vitro methods sometimes use pig ear skin due to its equivalence with human skin [24]. However, human skin models are the most generally accepted models for in vitro permeability tests [22].
These are methods that make use of computational models, most commonly, predicting drug absorption in the intestines [6]. Models only provide a guide on relative scales and do not necessarily reflect exact values that can be obtained from in vivo studies. These predictive permeability tools are essential for drug compound synthesis and manipulation of a drug template to improve absorption of the drug [25]. Caution should be taken by the user to understand the drug scaffolds and how best the software being chosen can fit in their work before purchasing. Literature reviews, user reviews and instincts can be good enough for a good choice of in silico tools. Commercial software and products for the determination of permeability are available with highly acceptable correlations to the reliable Franz cell data [26,27].
There are three common in vitro permeability assays in use; cell layer, Parallel Artificial Membrane Permeability Assay (PAMPA) and ‘Immobilized Artificial Membrane’ High Performance Liquid Chromatography (IAM-HPLC) [6]. All of these involve test sample partitioning into lipophilicity and aqueous phases.
Cell layer permeability method: This is the earliest method there was in the determination of permeability in drug discovery and covers models of passive diffusion, paracellular permeability, and active uptake transport and efflux mechanisms [6]. It models the permeability barrier of the epithelial membrane that drug compounds encounter in the duodenum, jejunum and the ileum, with Caco-2 and the Madin Darby Canine Kidney (MDCK) as the most commonly known cell lines used in this assay [28]. MDCK has been used for passive diffusion permeability predictions in drug discovery [29].
Cells are plated in the cell culture insert, a holder part of a device, where they settle into a porous support. The cells reach confluence over a period of about 21 days with growth covering upto the surface of the support usually forming a mixture of both multilayers and monolayers. However, it is important to work with monolayers and no gaps should be available to avoid the sample from just rapidly slipping through without appropriate barrier required. As time passes, the cells on the support develop microvilli morphology on the top surface. This particular Caco-2 method becomes relatively more expensive due to the duration it takes. Faster commercial culture techniques (5 days) are available as alternatives, take caution to have full transporter functionality. On the other hand, MDCK cells reach confluence in a 3 to 4 day period. Once the experiment begins, replace the growth medium with saline (buffered) containing the test compound and glucose [30].
From this stage, two experimental methods are common. The buffered test compound is laid on the apical surface of the cell layer and on the basolateral surface; the buffer is placed without test compound. The test buffered compound diffuses through the cells from the apical surface of the cell layer to the basolateral compartment. Specific time points are selected at which aliquots are collected from the two compartments over 1 to 2 hour durations. Liquid Chromatography/ Mass Spectroscopy (LC/MS) or HPLC is used to determine the concentration of the test compound in each compartment. This is termed the Apical to Basolateral Experiment (Ap-Ba) and it provides the permeability values in the absorptive direction, modeling absorption in the Gastrointestinal (GI) area. This method is effectively and reliably predictive of in vivo absorption [30].
The other experiment is to exchange the positions of the solutions being placed at the basolateral and the apical surfaces of the cell layer. The target here is to determine the permeability of the compound by cell membrane transporters. This is called the Basolateral to Apical Experiment (Ba-Ap). Comparing the results from the two experiments, conclusion can be drawn. If permeability is the same in the Ba-Ap direction as in the Ap-Ba, then passive diffusion is the primary mode of permeation for the compound. If the two results are significantly different, then it is predicted that a membrane transporter will be required [6].
Immobilized artificial membrane-High performance liquid chromatography (IAM-HPLC): In this method, phospholipids are used to covalently bond to solid support in place of octadecyl groups as used in reverse-phase HPLC. The phospholipids are made of polar-head groups and side chains of aliphatics of the lipids and partitioning of the test compounds is between the phospholipid phase and the mobile aqueous phase. The phospholipid affinity is directly proportional to the chromatographic capacity factor, k, which is used to rank test compounds in an order that shows ranges of phospholipid affinity since affinity parameters correlate with permeation [6]. A standard with known permeability is always used for calibration of the systems [31]. The IAM-HPLC is sensitive to structural variations which make it easier for the system to separate the entities by its ability to adjust the retention time [32,33]. The use of the HPLC format makes this method easy and convenient to apply. IAM-HPLC systems use isocratic mobile phases traditionally providing for longer retention times for compounds that are highly lipophilic. It is easier to work with this method as it only requires little amount of test materials and there is no interference from impurities on the prediction of permeability [34,35].
Parallel artificial membrane permeability assay (PAMPA): The PAMPA tests only passive diffusion with a clearer mechanism of permeability, independent of others. Unlike living cells being used as barriers, long chain hydrocarbon-solubilized phospholipids such as egg lecithin and phosphotidyl choline are used [6,36]. The sample with the test compound is diluted using an aqueous buffer (pH 7.4) making what is known as a donor solution [22]. This solution (usually, 25 µg/mL) is put in a 96-well plate, where each well is called the donor. A porous-bottomed 96-well filter plate is put over the donor well plate, directly in contact with the donor solution. A phospholipid solution (1-2 µL) is place into the wells of the filter plate and let soak through to the bottom, forming an artificial barrier. On top of this artificial barrier is placed a blank buffer and these wells are known as acceptors [37]. Figure 1 is a schematic diagram representing how donor and acceptor wells are set up in PAMPA with their respective solutions and how the drug molecules appear after incubation. The environment at the interface between these two plates is maintained at constant humidity and temperature for any duration between 1 and 18 hours, subject to the validation protocols of the operating laboratory. After this duration, samples are collected from both the acceptor and donor wells and the concentration of the test compounds in each is determined using Liquid ChromatographyUltraviolet (LC/UV), Liquid Chromatography-Mass Spectroscopy (LC-MS) or a UV plate reading device with the donor solution that was never placed into the plates being used as a standard [38]. Permeability through this method is often known as the effective permeability (Pe) [37]. It has a very high correlation with human jejunal permeability approximately comparable to that provided by Caco-2 [38]. With appropriate variations of the pH of the buffers, several other environments can be simulated. Sometimes, the acceptor buffer may be neutral and the acceptor pH lower, simulating the GI tract [6].
Figure 1: The PAMPA set up before and after incubation.
Drug LogP and LogDx are measures of drug lipophilicity, the extent of partitioning between an aqueous and an organic environment in the body with LogP being general information where all molecules in the test compound are neutral and LogDx being at any specific pH (x) environment, where some part(s) of the compound molecules under test is(are) ionic [39,40]. The methods are the same in both LogP and LogDx.
Shake flask method: Partitioning experiment of the test compound in octanol and water gradients can be conducted at a large scale as well as at a titer plate scale for higher throughput [41]. A flask can be used at a larger scale and well plates at a titre scale which will just be named a reaction vial in this paper. The test molecular compound is dissolved in Dimethylsulfoxide (DMSO) and added to the reaction vial. Since DMSO can potentially react with the reacting species during the experiment, its volume should be small (<1% of aqueous volume). Octanol and water are then added to the reacting vial. The vial is then tightly sealed and shaken to thoroughly mix the analyte solution and the two solvent phases. After mixing, a small aliquot from the mixture is drawn and the concentration of the compound is determined by analysis usually using HPLC [42,43]. Figure 2 is a schematic diagram of a simple shake flask method in the determination of lipophilicity for a drug compound.
.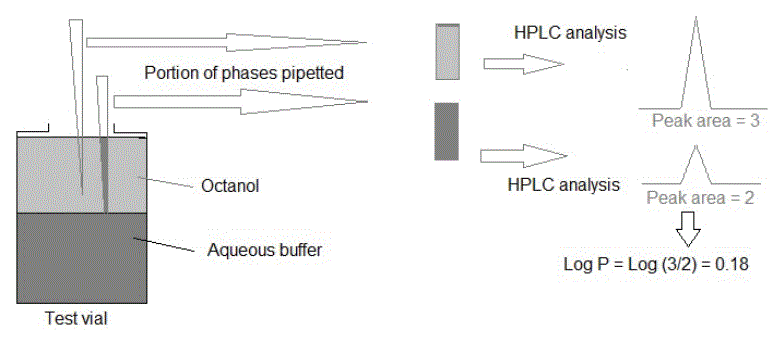
Figure 2: Shake flask lipophilicity determination method.
During the HPLC analysis, there is usually a challenge with carryover from one HPLC injection to the next. To overcome this problem, the HPLC system needs to be flushed after every run to avoid contamination with wastes from previous runs.
Reversed phase HPLC LogP and LogDx determination method: A typical multistage partitioning technique is used in this method where analytes are distributed into the aqueous mobile phase and the organic stationary phase of an HPLC column [44]. Standards whose lipophilicities are already known from other methods are first injected into the RP-HPLC column in a series. These series of standards are essential for the availability of enough data for the development of a calibration curve which is plotted from the times of retention of all the standards against their previously measured lipophilicity values [45]. Without changing HPLC conditions, the test compound is then run and its retention time compared with those on the standard calibration curve to determine its lipophilicity [46-48].
Capillary electrophoresis LogP and LogDx determination method: This method uses the Microemulsion Electrokinetic Chromatographic Techniques (MEEKC). Partitioning of the test compound is between an aqueous phase and a non-polar organic microemulsion phase. Standard calibration curves are also drawn as in the RP-HPLC method and the test compound retention time is compared to the calibration curve for lipophilicity determination [49-51].
pH-Metric method: This method basically employs titration techniques to determine liphophilicity [52]. An acid or a base is used as a titrant with the test compound as an analyte to draw a titration curve, for convenience called A in this paper. This experiment is then repeated, this time with 23% octanol added to the test compound and the titration curve is drawn again, for convenience, called B in this paper. A shift in the titration curve from A to B indicates the degree of partitioning into octanol, for the test compound which provides the basis for the calculation of lipophilicity [52].
Various chemical data bases are available with LogP and LogDx data for compounds analyzed previously. These past analysis results provide validation tools for the improvement of the algorithms in use or the development of a new algorithm. One of the mostly used in silico method is the fragment method [53]. In this method, logP and LogDx values of substructures are determined and stored in the database. Any new molecule is then broken down into matching sub structures whose individual contributions are summed up to determine logP and LogDx values for the new molecule. It should be noted that these in silico results are not usually expected to be the same as laboratory values; they have an average difference of about 1.05 log units [6,54]. It is therefore imperative to report the method used to obtain any reported lipophilicity values. However, the differences are very small when predicting trends in lipophilicity, making in silico tools more predictive in this use. The advantages of in silico methods include the ease of access and operation without need for various solvent systems and worry about compound solubility and impurities. The cost for the determination of lipophilicity in silico is much lower than experimental determinations. They are rich in the diversity of compounds and other drug-like properties apart from lipophilicity.
Drug pKa , permeability and lipophilicity values are essential to drug development as they directly impact on the bioavailability of the drug molecule to the intended biological site of action. It is noted that drug-like property values are not always the same when different methods for the same assay are used. Another detail of note is that though critically important for botanic-based drug development, these tests are usually ignored, letting consumers use without knowing whether the tested bioactive compound is bioavailable to the biological target or not, This often wastes time and other resources if it is found that the active compounds are not bio-available in the body due to poor drug molecular-structural properties. It is high time that various analytical methods to be put forward and help Structure Property Relationships (SPR) be treated as equally important as Structure Activity Relationships (SAR) in drug discovery, designing and development to save resources.
When reporting results for these tests, it is always imperative to mention the method and conditions used as these are largely predictive and may differ based on methods taken and conditions thereof. It is also noted that models for most of these tests are also limited to a few organs and membranes. There is need for the development of more methods that can encompass wider organs as reaction sites. It is important for drug discoverers, designers, manufactures, as well as funders to always consider “drug-like” property determination on their protocols at an earlier stage to only focus on the best promising leads and minimize resource wastage on poor molecules, a decision of which to modify would be made at this early stage.
The limitations to this work include the omission of details about commercially available quick test tools. However, this paper has covered the fundamentals from which modern techniques derive their methods.
The authors acknowledge PHARMBIOTRAC ACE-II project of Mbarara University of Science and Technology for funding this work.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Mtewa AG, Ngwira K, Lampiao F, Weisheit A, Tolo CU, et al. (2018) Fundamental Methods in Drug Permeability, pKa , LogP and LogDx Determination. J Drug Res Dev 5(1): dx.doi.org/10.16966/2470-1009.146
Copyright: © 2018 Mtewa AG, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access