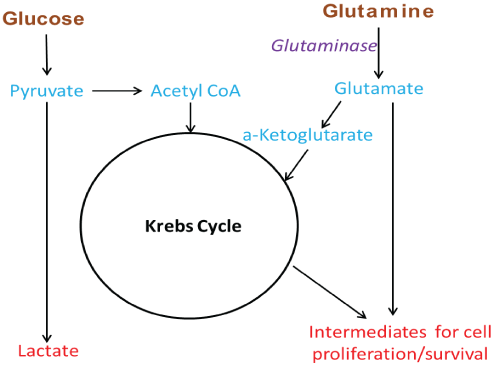
Figure 1: Simplified diagram of glutamine use in Kreb cycle anaplerosis and other intermediate production

Lee McDermott*
Department of Pharmaceutical Sciences, University of Pittsburgh, 3501 Terrace Street, 808 Salk Hall, Pittsburgh PA 15261, USA*Corresponding author: Lee McDermott, Department of Pharmaceutical Sciences, University of Pittsburgh, 3501 Terrace Street, 808 Salk Hall, Pittsburgh PA 15261, USA, Tel: 412-648-9706; E-mail: lam179@pitt.edu
A number of tumor cell lines exhibit dependence on glutamine in vitro, a phenomenon termed as ‘glutamine addiction’. The rate limiting step in the processing of glutamine in mitochondria is controlled by glutaminase, an amidohydrolase that converts glutamine to glutamate. In many tumor cell lines the kindney-type glutaminase (KGA) and particularly its C isoform (GAC) are upreglulated. As such, inhibition of KGA/GAC has been viewed as an attractive strategy for exploiting tumor cells’ glutamine addiction for cancer therapy, as it aims to deprive them from their ability to metabolize a nutrient they apparently need for their survival and proliferation. In this very brief review we discuss the progress toward the identification of KGA/GAC inhibitors.
Cancer; Glutaminase inhibitors; KGA; GLS1; GAC; LGA; GLS2; BPTES; Compound 968; CB-839
Metabolic differences between normal and cancer cells were observed for the first time about ninety years ago by Otto Warburg. In his 1924 and 1925 papers ‘Ueber den stoffwechel der tumoren’ (About the metabolism of tumors) and ‘Ueber den stoffwechsel der carcinomzelle’ (About the metabolism of the cancer cell) Warburg was the first to show that when compared to normal tissue/cells, tumors exhibit increased rate of glucose uptake and lactic acid production even when ample oxygen was available [1,2]. Thirty-one years after the report of this phenomenon, called aerobic glycolysis or the Warburg effect, in 1955, Harry Eagle in his paper ‘Nutrition needs of mammalian culture cells’ described another interesting observation. HeLa cancer cells have high glutamine requirements in vitro for optimal growth when compared to normal mouse L fibroblasts [3]. Since then, research on the role of glutamine in cancer cell proliferation has revealed that high glutamine utilization and dependence, a property termed as ‘glutamine addiction’, is a metabolic hallmark of many tumor cells in vitro [4,5]. The Warburg effect has been linked, to the dysregulation of a number of pathways known for their involvement in cancer and multiple theories have been advanced for explaining its biological benefit to cancer cells [6,7]. Nevertheless, despite considerable progress in understanding this phenomenon, success in exploiting it for cancer therapy has thus far been limited to tumor imaging (18FDG-PET) [8]. Glutamine addiction, like the Warburg effect, has also been linked to dysregulation of pathways involved in cancer. The biological rationale proposed for it lies on the fact that, as glucose is quickly uptaken and converted to lactate, tumor cells increase their use/uptake of glutamine to produce a-ketoglutarate, to anapleroticaly fuel their Krebs cycle, and to also produce intermediates for the synthesis of lipids, nucleosides and other biomolecules needed for their proliferation and survival (Figure 1) [4,5]. As many cancer cells appear addicted to glutamine, taking advantage of this addiction has been considered as a very attractive strategy for cancer treatment and has drawn much attention, particularly over the last 10 years.
Figure 1: Simplified diagram of glutamine use in Kreb cycle anaplerosis and other intermediate production
Glutaminase is an amidohydrolase that converts glutamine to glutamate in the first step of the glutamine processing in mitochondria. The human genome encodes two main glutaminase isoforms, the kidney isoform (KGA/GLS1) and the liver isoform (LGA/GLS2). LGA is encoded by the GLS2 gene in chromosome 12, and it is highly expressed in liver and to a much lower degree in brain and pancreas [9]. KGA is encoded by the GLS gene, in chromosome 2, and has much wider tissue distribution than LGA [9]. Both enzymes are catalytically active as tetramers but have very different kinetic behavior in terms of activation by inorganic phosphate, Km for glutamine, and/or inhibition by glutamate [10]. In the 1969 paper ‘The proportionality of glutaminase content to growth rate and morphology of rat neoplasms’, Knox et al. [11] demonstrated that kidney glutaminase activity is proportional to the growth rate of tumors in rats. Since that paper, cumulative evidence suggests that KGA, and in particular the kidney glutaminase isoform C (GAC) splice variant that has a 71 residue shorter c-terminus, could be an important target for therapy [12-14]. TCGA (The cancer genome atlas) data suggest that there is high GLS gene expression in a number of cancers. Upregulation of KGA, and particularly GAC, has been seen in a number of human tumor cell lines and correlates with increased proliferative rates and in certain instances with tumor progression [5,13,15-19]. The observed KGA/GAC upregulation has also direct links to transcription factors and enzymes/pathways with concrete involvement in cancer such as Myc, STAT-1, c-Jun, Erb2 and the RAF-RAS-MEK-ERK signaling pathways [20-25]. Furthermore, inhibition of KGA expression has been shown to inhibit tumor cell growth [26]. In this context, impairing the ability of tumors to process glutamine by selectively inhibiting KGA/GAC with small molecules has been viewed as an appealing and likely viable strategy for anti-cancer therapy.
A review of the literature suggests that DON (Figure 2) and L-2- amino-4-oxo-5-chloropentanoic acid, are the earliest compounds cited as KGA inhibitors [27]. Both compounds act as glutamine mimics and inactivate the enzyme by covalently binding in the active site. Neither is KGA selective however, and both interact with several other targets in addition to glutaminase [28-30]. Among the two, DON also made it to the clinic but its development was halted due to excessive toxicity, apparently the result of its broad enzyme inhibition spectrum [31,32]. A screen of the 1280 member Library of Pharmacologically Active Compounds (LOPAC1280) identified ebselen, apomorphine and chelerytrine as agents that could inhibit KGA/GAC (Figure 3).Unfortunately the utility of these agents in studies/therapy is limited because they interact with other targets and their KGA/GAC vs LGA selectivity is very narrow [33]. In a different screen, thiourea THDP-17 (Figure 3) was identified as an un-competitive KGA inhibitor [34]. KGA/GAC vs LGA selectivity data have not been reported for this agent. Compound 968 (Figure 3) identified as KGA/ GAC inhibitor after protein pull down experiments where lysates from Cdc42-(F28L)-expressing NIH 3T3 cells were incubated with streptavidin beads labeled with the biotinylated N,N-dimethyl-bromophehyl moiety of 968, a key moiety for its activity [35,36]. Studies suggest that 968 acts allosterically and preferentially binds to GAC monomers [37]. The KGA/GAC vs LGA selectivity of this compound has not been reported. However, there are data suggesting that 968 may also bind LGA [38]. As a probe, compound 968 has been featured in a number of biological studies. It has been shown to afford growth inhibition in glutamine-addicted cancer cell lines as well as growth inhibition in vivo in a P493B lymphoma mouse model [35,36,39-43]. The first truly selective KGA/GAC inhibitor reported was BPTES/SNX-1770 (Figure 4). This compound, along with a small list of close derivatives, first appeared in patent application US 2002/0115698 A1. Initial kinetic and biophysical studies showed that the compound is a highly selective allosteric KGA inhibitor that upon binding leads to formation of inactive KGA tetramers [44]. Crystallographic work, using KGA and GAC, confirmed the BPTES allosteric binding mode and showed that the compound binds at the interface between two interacting KGA/GAC dimers, in a 2:4 stoichiometry, and stabilizes an important flexible loop between residues Glu320-Pro327 near the active site (Figure 4) [24,45,46]. In the binding region of BPTES, KGA/GAC and LGA differ in only 2 amino acids. Specifically, Phe318 and Phe322 in KGA/GAC are substituted in LGA by Tyr251 and Ser255 respectively. These differences and particularly the Phe322/Ser255 difference are key for the BPTES binding to KGA/GAC. Studies with KGA/GAC mutants revealed that a Phe318/Tyr Phe322/Ser double mutant or a KGA Phe322/Ser single mutant were unable to bind BPTES but they retained catalytic activity, while a Phe318/Tyr single mutant was catalytically active and also bound BPTES [24,45]. BPTES has been shown to effect cell growth inhibition in glutamine addicted cancer cell lines in vitro and tumor growth inhibition in test animals [19,22,39,47-52]. Despite of its demonstrated activity BPTES did not advance to the clinic because of poor drug-like properties [52]. Preclinical formulation work that aimed to overcome its poor properties was recently published and demonstrated that BPTES encapsulation in poly (lactic-coglycolic acid)-poly (ethylene glycol) (PLGA-PEG) nanoparticles improved compound delivery and tumor uptake and afforded improved tumor growth inhibition than un-encapsulated BPTES in a JH090 pancreatic tumor model [52]. Medicinal chemistry efforts aiming to expand the SAR (structure activity relationships) around BPTES and also produce BPTES derivatives or analogs/mimics with improved potency and drug-like properties have been reported by our laboratory and others and selected compounds from these efforts are shown in (Figure 5) [53-56]. BPTES derivatives and BPTES analogs/mimics have also been claimed in recently published patent applications [57-65]. Among the broader class of BPTES-like compounds, the most advanced is CB-839 (Figure 6) [57,66]. This compound is a more potent KGA/GAC inhibitor than BPTES. It binds in a similar fashion, was shown to have activity against various cancer cell lines and to effect tumor growth inhibition in the CTG-0052, JIMT-1, Caki-1 and other tumor models [18,56,66-69]. CB-839 is currently in Phase I/II clinical trials in combination with other agents (NCT02071927, NCT02071888, NCT02861300, NCT02771626, NCT02071862, NCT03047993 and NCT03057600). As a single agent CB-839 appears to exhibit an acceptable safety profile despite the relatively high, 600 mg twice a day with food, dose regimen required for maintenance of sustained therapeutic plasma levels and for the alleviation of its high absorption variability [70,71]. Early clinical reports suggest that the CB-839/azacytine combination for acute myeloid leukemia (AML), the CB-839 combination with everolimus for renal cell carcinoma (RCC), the CB-839/pomalidomide and dexamethasone combination for multiple myeloma (MM) and the CB-839/paclitaxel combination for triple negative breast cancer, warrant further study [72-75].
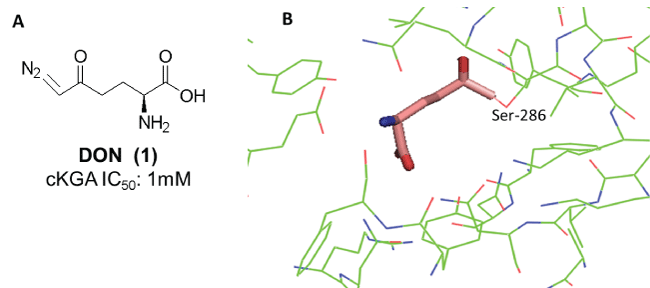
Figure 2: A: Structure of DON and its activity against the catalytic domain of KGA (cKGA) [30]. B: cKGA-DON co-crystal (PDB: 3VOY) showing covalent attachment to Ser 286 in the active site
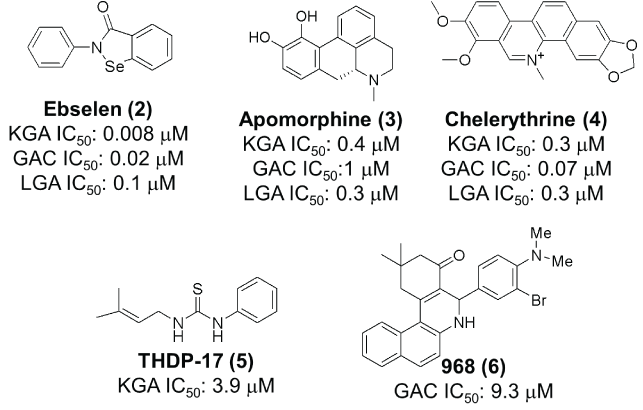
Figure 3: Structures of ebselen, apomorphine, chelerythrine , THDP-17 and 968 and their reported activities [33,34 and 36 respectively].
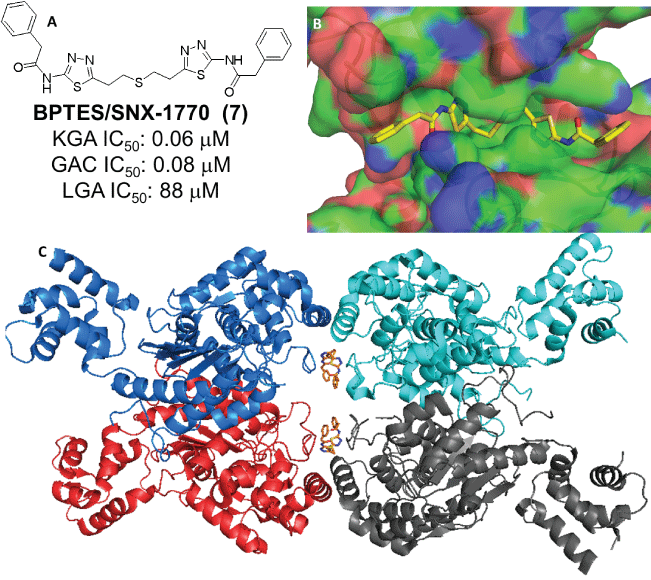
Figure 4: A: Structure of BPTES and its glutaminase selectivity profile [45]. B & C: Crystal structure of BPTES in complex with GAC (PDB: 3UO9)
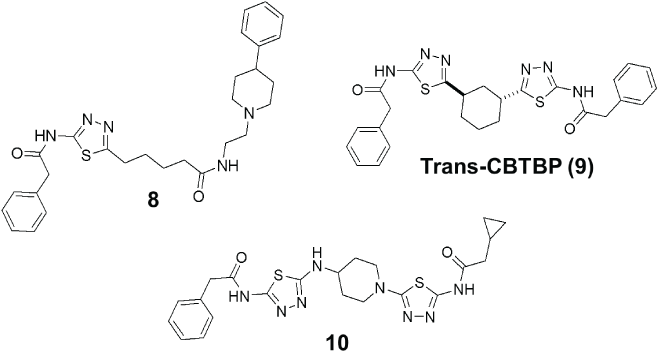
Figure 5: Selected examples from reported medicinal chemistry efforts aimed to improve upon BPTES
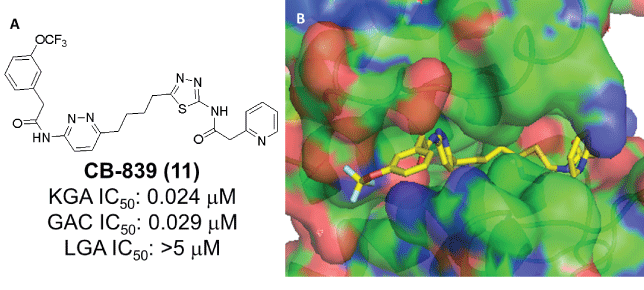
Figure 6: A: Structure of CB-839 and its glutaminase selectivity profile [52,53]. B: Crystal structure of CB-839 in complex with GAC (PDB: 5HL1)
The ability of selective KGA/GAC inhibitors to potently inhibit the growth of glutamine addicted cancer cells and afford tumor growth inhibition in vivo is well documented. The preclinical in vivo experience with these compounds, however, suggests that, as single agents, they can only achieve tumor growth inhibition and not tumor regression or stasis, as one would expect given the attributed importance of glutamine in Krebs cycle anaplerosis, the synthesis of nucleosides and other intermediates important for cell survival and proliferation [35,44,53,62,66,69,76,77]. The inability of KGA/GAC inhibitors to afford tumor stasis or regression as single agents may be accounted for by the fact that tumors are heterogeneous, they can use alternative fuels, such as acetate and lipids, and recent findings which show that the metabolic behavior of tumor cells in vitro and in vivo can be vastly different [78- 83]. Taking all of this into consideration it seems likely that, in cancer therapy, KGA/GAC inhibitors will be most useful in combination with other agents rather than as a stand-alone treatment. Preclinical exploration of combination regimens of KGA/GAC inhibitors with established chemotherapeutic agents and/or inhibitors of complementary pathways suggests that synergy is indeed possible [41,43,62,66,69,84-92]. For instance, the available preclinical in vivo data with CB-839 show that combinations with everolimus, paclitaxel, pomalidomide, 5-FU, 5-AZA, and anti-PD-L1/anti-PD-1 antibodies have enhanced efficacy and can produce tumor stasis/regression [66,69,85,90,92,93]. This offers hope that such combinations may translate to the clinic. Early clinical reports in that front appear encouraging [72-75]. However, it is too early to proclaim success. Ongoing and future clinical trials are expected to shed more light on the utility of these regimens and the utility of KGA/GAC inhibitors in general for cancer treatment.
Download Provisional PDF Here
Article Type: Mini Review
Citation: McDermott L (2017) Kidney-Type Glutaminase Inhibitors for Treating Cancer. An Overview. J Drug Res Dev 3(2): doi http://dx.doi.org/10.16966/2470-1009.133
Copyright: © 2017 McDermott L, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access