Abstract
Here, we demonstrate how array-based label-free biosensors can be applied for high-throughput, high-information epitope mapping of monoclonal antibodies (mAbs). High-resolution epitope mappingusing antigenic variants enable the identification of specific binding regions on Abs. The use of epitope mapping can expedite the discovery of therapeutic mAbs and vaccines and inform the number and diversity of the interactions being studied. Specifically, we show the opportunity epitope mapping offers in distinguishing subtle differences for target-engagement amongst otherwise highly similar mAbs.
Keywords
Monoclonal antibodies; Epitope mapping; Label-free biosensors; High-throughput screening
Substantial Value Exists in Characterizing How
Antibodies Engage Antigen
Therapeutic mAbs are now FDA approved for clinical indications of most major disease classes including cancer, infectious, autoimmune, cardiovascular, pulmonary, and inflammatory disease [1] and have a market value of over $50 billion accounting for seven of the top ten selling drugs in sales [2,3]. An extremely competitive landscape exists for the development of new therapeutic mAbs as many targets require sophisticated approaches to identify and develop candidates that target functional epitopes or are superior to a competitor molecule(s) [1,4]. However, selection criteria in traditional screening approaches fail to account for intellectual freedom to operate. Consequently, therapeutics developed against high-profile targets can find substantial challenges at the commercialization stage. Additionally, the sequence and structural variability of many targets also presents challenges in identifying a lead candidate with broad activity. This is especially true for highly mutative targets found in oncology. A desirable drug candidate is not easily perturbed by modifications in a functionally relevant target epitope, which can occur for instance during different disease states or treatment regimens. Similarly, development of efficacious vaccines requires eliciting immune responses capable of targeting regions that often can demonstrate sequence hypervariability. Understanding immune responses elicited toward specific aspects of a viral target is critical in the development of treatments for infectious disease. The enhanced techniques for epitope mapping described here provide the capability to improve the discovery workflow for the next generation of therapeutics.
Epitope Mapping Provides Many Advantages for Therapeutic
Screening
Epitope-based screening is more advantageous than traditional affinity-based screening for selecting therapeutic mAb candidates [5-8]. Furthermore, when combined with functional assays, epitope based screening identifies candidates to functional epitopes leading to fewer dead end candidates increasing the probability of success in Phase II and III trials [9]. In combination, epitope binning and epitope mapping using label-free biosensors can thus increase the probability of success, reduce overall costs, and enhance R&D productivity [9]. Competitive epitope binning is best suited as a primary stage screening tool to cluster mAbs based on sites of epitope engagement. When conducted using labelfree biosensing, such as array-based surface plasmon-resonance imaging (SPRi), the assay only requires crude or purified mAbs and the antigen they are raised against. While defining epitope bins is an important first step in candidate selection, it does not directly localize the epitopes on structure nor does it typically involve screening against more than one form of antigen.
To localize epitopes and understand the impact on variations in target sequence and structure, epitope mapping is an excellent tool [10]. Epitope mapping via array SPRi can be conducted in primarily two ways (1) an overlapping library of antigen peptides is arrayed across the surface and probed for binding to mAb injections or (2) target variants/mutants are injected across an array of mAbs coupled to a biosensing surface [11]. The library, also known as a reference panel, approach is advantageous for quickly mapping sites of binding for mAbs recognizing linear epitopes; it typically does not contain sequences with higher order structure, so that conformational binders are not mapped. Depending on objectives, this information may be sufficient for defining structural-spatial relationships of epitope binning communities. The epitope mapping approach using mutants/variants does require cloning, expression, and purification of each variant in question, but binding to conformationally sensitive mAbs can be monitored. Additionally, the mutant/variant approach lends itself to comparative screening approaches against multiple antigen isoforms for instance. In both formats, the real-time nature of the data collection affords the opportunity to understand the rates at which interactions occur, providing further understanding of the interaction. In other screening approaches, such as ELISA, the lack of real time data collection can be problematic, for instance, when binding is weak. Here, we present an epitope mapping approach that can both enhance the structural characterization of an antigen and its relationship against mAbs. Epitope mapping was done using high-throughput SPRi with multiple mutant constructs of herpes simplex viral glycoprotein D(gD) and a panel of mAbs with broad epitopic coverage of gD. In a single experiment, this approach confirmed existing mapping data while detecting new binding profiles not previously identified. These results highlight the power of epitope mapping to quickly screenantigen:mAb interactions for a large panels of mAbs and generate a vast amount of information to drive vaccine and therapeutic drug development.
Materials and Methods
Reagents
Recombinant, mutant HSV gD constructs were produced from baculovirus-infected insect (Sf9) cells as described previously [12-15]. The extracellular portion of gD, where antibody responses would be relevant from a therapeutic perspective, was used as the starting point for establishing mutants. In brief, mutants were obtained by expression of N-and/or C-terminus truncated species, insertion or deletion of novel sequence within the wild type sequence to modify protein structure, or else swapping of individual residues to identify those critical for mAb binding. Mouse monoclonal mAbs raised against HSV gD Type 1 or Type 2 were obtained from several sources and are described in previous manuscripts, including their canonical grouping assignments based on lower through put mutation, mapping, and competition studies [16,17].
Array SPRi
An array of approximately 40 anti-HSV gDmAbs was coupled onto a biosensor surface using a Continuous Flow Microspotter (CFM). In brief, covalent attachment of the mAbs to the surface was done by first activating the carboxymethylated dextran surface with a mixture of 1-Ethyl-3-(3- dimethylaminopropyl) carbodiimide (EDC) and n-hydroxysuccinimide (NHS). MAbs prepared in sodium acetate pH 4.0 were then flowed over discrete locations on the array to allow for coupling via amine groups. Remaining active esters were quenched using ethanolamine. To investigate regions of sequence key for mAb binding, gD mutants were injected across the mAb array and detected using an IBIS MX96 SPR imager, which monitors the presence of mass binding to each of the 40 mAb locations on the biosensor surface in real time. Due to inactivation during the coupling procedure or else non-reactivity towards the subtypes of gD used in this study, some mAbs did produce binding signals in this assay (data not shown). Receptors to gD, Nectin-1 and HVEM, were injected following binding of gD mutants to the mAb panel to assess whether mAbs could disrupt receptor binding. Bound components were removed from the mAb surfaces by injection of glycine pH 2.0 after each cycle.
Results
Localization to the sites of gD bound by specific mAbs was investigated using variants of gD, Figure 1. The canonical group assignments of these mAbs, based on previous studies, is listed at the top of the figure, along with the ability of mAbs to compete with receptors Nectin-1 and HVEM for binding to extracellular gD. Receptor competition largely followed the canonical group assignments, consistent with what is known about these mAbs from a structural binding perspective.

Figure 1. Epitope mapping of mAbs to mutants of gD. MAbs without previous canonical assignment are listed as (Unk). Disruption of Nectin-1/HVEM binding is shown as (+) and mAbs not tested for receptor blocking are not determined (ND). Red cells indicate lack of binding to mutants, yellow cells indicate low levels of binding, and green cells indicate high binding responses. The inset displays SPR sensor gram signals, as response units (RU) over time (seconds).
Binding responses towards mutants by anti-gD mAbs are represented as a heat map, Figure 1. Mutants of gD are listed as rows and mAbs are arranged as columns. Green cells denote strong binding signals, yellow cells are intermediate binding signals, and red cells identify lack of binding. The variants not only localize binding to N- or C-terminal regions of gD, but also emphasize specific residues that are critical for binding. Referring to the canonical Group Ia and Group Ib mAbs, mutants Y38A (306t) and V231W (306t) can be used to differentiate these sub groups. They demonstrate that although these mAbs have many shared traits that have led to their canonical grouping (e.g. binding despite the V231W mutation), specific residues critical for binding can be used to distinguish them with an epitope mapping approach (Y38A) as shown in Figure 2. Furthermore, the throughput capabilities of this approach identified mAbs that appear similarly sensitive to the Y38A mutation and previously had not been mapped, such as 108S and 77S. Sensorgram profiles are shown in an expanded view for representative mAbs from Group Ia and Group Ib to highlight the differences in binding responses observed and emphasize the value this SPRi approach offers by tracking binding in real time. The Y38A mutation disrupted nearly all binding of the Group Ib mAbs but not the Group Ia mAbs. Receptor competition profiles and epitope mapping profiles are consistent with the structure of gD (see Figure 2 and Supplementary Video).
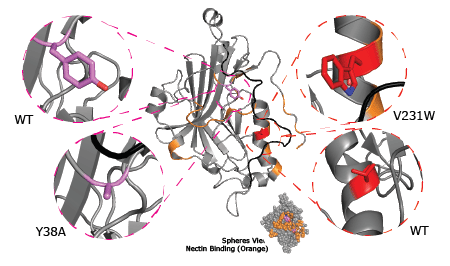
Figure 2. 3D structural representation of the two mutationsof glycoprotein Dshown in figure 1. Crystal structure (pdb model 2C36) of gD. Mutations are shown in greater detail to highlight the structural difference identified in this epitope mapping study.
Discussion
High-throughput SPRi on a panel of anti-gD mAbs by epitope mapping using mutants confirmed previous epitope relationship mappings as well as identified binding regions for several mAbs that were previously undetermined. While epitope mapping can be performed with ELISA, the real-time, label-free detection is much more information rich (Figures 1, 2). Additionally, array based SPRi also has advantages compared with ELISA in that each injection of variant sees all unique mAb locations on the array simultaneously, reducing overall antigen requirements. Challenges with mutant epitope mapping come with the need to clone, express, and purify a large panel of antigen variants. Commonly, alanine scanning (i.e. shotgun mutagenesis) [18] is used to create an epitope mapping reference panel where individual residues are changed to alanine and the binding properties are assessed [18-20]. Resource requirements have limited adoption of this approach; however, with improved platforms for generation of these panels [11] and improved technologies, including array-based SPRi for high-throughput screening, this workflow can generate information that was not previously available early in discovery. Opportunities to enhance modeling of antigen and antibody relationships also exist using the epitope mapping approach. This information derived from these types of studies is well suited for informing protein docking prediction algorithms. Additionally, epitope mapping in conjunction with structure and binding analysis (biophysical prediction software) could improve mutagenic design and layout.
Conclusions
The results presented in this manuscript show how epitope mapping with conformational mutants is a versatile and robust tool to expand the screening repertoire in drug discovery and vaccine development. In conjunction with other epitope characterization approaches such as competitive binning and receptor competition, a detailed picture of antigen/antibody relationships can be developed. A key enabler to this workflow is the use of real-time biosensing, demonstrated here using array-based SPRi, that can efficiently accumulate data in a multiplexed fashion and present a complete picture of the binding interactions in question. Collectively, the data from these studies can enable new developments in predictive modeling and ultimately improve drug and vaccine development.
Article Information
Article Type: Research Article
Citation: Ditto N T, Cairns TM, Lou H, Closmore A, Eisenberg RJ, et al. (2016) Understanding antibody: Antigen Relationships using Antigenic Variants with Array-Based Spriepitope Mapping. J Drug Res Dev 2(4): doi http://dx.doi.org/10.16966/2470-1009.124
Copyright: © 2016 Ditto N T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 08 Nov 2016
Accepted date: 15 Dec 2016
Published date: 20 Dec 2016
