Abstract
Background: Androgen deprivation therapy (ADT) by surgical or medical castration is recommended for advanced or metastatic prostate
cancer. Recent literature suggests that medical castration by luteinizing hormone receptor hormone (LHRH) antagonists might have advantages
over treatment with LHRH agonists in patients with metastatic prostate cancer when prostate specific antigen (PSA) progression free survival and
overall survival are concerned. Using a state-of-the-art method to assess levels of testosterone, we investigated whether a potential difference
in clinical outcome between different forms of ADT might be related to differences in serum testosterone concentrations. We further searched for
evidence in literature for other biochemical pathways explaining a potential benefit of LHRH antagonists over LHRH agonists.
Methods: Patients underwent surgical castration (n=34) or received an LHRH antagonist (n=25). Serum samples were obtained more that
3 months after initiation of ADT. Testosterone levels were determined using isotope dilution-liquid chromatography tandem mass spectrometry.
Dehydroepiandrosterone sulphate (DHEAS), androstenedione, sex hormone-binding globulin (SHBG) and inhibin B levels were determined.
Results: All surgically castrated subjects and all but one subject in the LHRH antagonist group had serum testosterone values less than 50
ng/dL. No difference was found between groups in serum testosterone, DHEAS, androstenedione and SHBG. Patients who underwent surgical
castration had significantly lower levels of inhibin B compared to patients treated with degarelix
Conclusion: Using a highly sensitive and specific technique of testosterone determination, no difference was found between patients after
surgical castration and patients on LHRH antagonists. Thus, differences in clinical outcome between different forms of ADT are accounted for by
testosterone independent pathways or mechanisms.
Abbreviations
ADT: Androgen Deprivation Therapy; CV: Coefficient of Variation; DHEAS: Dehydroepiandrosterone Sulphate; EGF:
Epidermal Growth Factor; EGFR: Epidermal Growth Factor Receptor; FSH: Follicle Stimulating Hormone; ID-LC-MS/MS: Isotope Dilution-Liquid
Chromatography-Tandem Mass Spectrometry; LH: Luteinizing Hormone; LHRH: Luteinizing Hormone-Releasing Hormone; LHRH-R: Luteinizing
Hormone-Releasing Hormone Receptor; LOQ: Limit Of Quantification; LUTS: Lower Urinary Tract Symptoms; PSA: Prostate-Specific Antigen;
RIA: Radio Immuno Assay; s.c.: Subcutaneously; SHBG: Sex Hormone-Binding Globulin.
Introduction
Androgen deprivation therapy (ADT) by either bilateral orchiectomy
(surgical castration) or medical castration (luteinizing hormonereleasing
hormone (LHRH) agonists, LHRH antagonists or estrogens) is
recommended for advanced or metastatic prostate cancer [1]. The aim
of ADT is to reduce serum testosterone concentrations to a castrate level
which is currently defined as <50 ng/dL, although recent developments
advocate for lowering this threshold to <20 ng/dL [2,3].
LHRH agonist therapy results in an initial increase in serum testosterone
concentration, also known as flare or flare-up. Anti-androgens can be
administered to counteract the symptoms of this initial rise in serum
testosterone, but at present, there is a lack of solid evidence for its clinical
necessity [4]. The LHRH antagonist degarelix (Firmagon®) has shown to be
non-inferior to LHRH agonist treatment at maintaining low testosterone
levels in patients with metastatic prostate cancer [5]. A recent study has
pooled the results of five randomized trials comparing LHRH antagonists
with LHRH agonists. The authors concluded that degarelix was associated
with prostate-specific antigen (PSA) progression-free and overall survival
compared with LHRH agonists [6].
Other studies showed that treatment with degarelix leads to greater
reductions in serum alkaline phosphatase levels in patients with
metastatic prostate cancer compared to leuprolide over a 1-year treatment
period [7]. Also, it was shown that degarelix might improve lower urinary
tract symptoms (LUTS) and achieves a greater reduction in prostate
volume in prostate cancer patients compared to goserelin combined with
bicalutamide [8-10]. In men with preexisting cardiovascular disease,
LHRH antagonists appear to reduce the number of cardiac events during
the first year of treatment compared to LHRH agonists [11].
Recent evidence suggests that an association is present between levels
of serum testosterone in men on ADT and clinical outcome. Progressionfree
survival and cancer–specific survival are reported to be higher in
those on ADT with sustained low testosterone levels compared to those on
ADT who experience testosterone breakthroughs of 32-50 ng/dL [12,13].
In five different studies that compared the activity of degarelix to a LHRH
agonist (i.e. leuprolide or goserelin) in patients with metastatic prostate
cancer, those receiving degarelix showed a significant lower risk of PSA
progression or death in the first year of treatment [6]. For now, the exact
explanation for this difference is unknown, but it might well is that more
thorough and sustained suppression of serum testosterone levels might be
one of underlying mechanisms [7].
In previous comparative studies, measurements of serum testosterone
levels were done by poorly performing immunoassays, making definite
conclusions on the timing to achieve a castrate level of serum testosterone
and the levels of serum testosterone themselves hardly possible [14]. In
this study, we describe the results of serum testosterone measurements
in patients with advanced or metastatic prostate cancer on LHRH
antagonist therapy using a highly sensitive and specific isotope dilutionliquid
chromatography-tandem mass spectrometry (ID-LC-MS/MS)
method [15]. We compared the testosterone concentrations in patients
on degarelix to those in surgically castrated men. Using a state-of-the-art
method to assess levels of testosterone, we investigated whether a potential
difference in clinical outcome between different forms of ADT might be
related to differences in serum testosterone concentrations. We further
searched for evidence in literature for other biochemical pathways and
mechanisms explaining a potential difference between LHRH antagonists
and LHRH agonists.
Materials and Methods
Study population
In this retrospective study, a total of 59 subjects were included. Thirtyfour
patients underwent surgical castration, 24 because of advanced or
metastatic prostate cancer and 10 patients as part of a gender transition.
There were 25 patients who received degarelix for metastatic prostate
cancer at a starting dose of 240 mg subcutaneously (s.c) for 1 month,
followed by s.c. maintenance doses of 80 mg monthly. None of the patients
received other hormonal therapies such as anti-androgens, abiraterone,
enzalutamide, ketoconazol or any other medication that could interfere
with the gonadal axis. All patients were treated for at least three months
before blood samples were drawn.
Serum testosterone determination
Venous blood was collected at a random time during the day from each
subject. The day of venous blood sampling was at least one week after the
last degarelix injection and at least one week before the next scheduled
LHRH antagonist injection. Serum was aliquoted and stored at -20°
Celcius until assayed.
Serum total testosterone was measured using the ID-LC-MS/MS as
described in detail before [15]. The lower limit of quantification (LOQ)
was 0.1 nmol/L (or 2.9 ng/dL), intra-assay and inter-assay coefficient of
variation (CV) at levels less than 1,0 nmol/L were less than 5% and less
than 13%, respectively.
Other parameters
Androstenedione was measured by a radio immuno assay (RIA) (DSL,
Webster, Texas) which featured a LOQ of 0.5 nmol/l. Intra-assay and interassay
CV for levels greater than 6 nmol/L were 6% and 9%, respectively,
and for levels less than 6 nmol/L were 8% and 12%, respectively. RIA
was also used for dehydroepiandrosterone sulphate (DHEAS). The LOQ
was 0.2 µmol/L. Intra-assay and inter-assay variation at 3 µmol/l was 6%
and 10%, respectively, and at 10 µmol/l was 4% and 9%, respectively.
An immunometric assay on an Immulite® 2500 (Siemens Diagnostics)
was used to determine the sex hormone-binding globulin (SHBG)
concentration. The LOQ for SHBG was 2 nmol/l, and the intra-assay and
inter-assay CV for the whole range was less than 3% and 4%, respectively.
Inhibin B was measured using a immunoassay (Beckman Coulter). The
LOQ was 15 ng/L. Intra-assay and inter-assay CV for the whole range was
less than 3% and 4%, respectively.
Statistics
Statistical analysis was done using SPSS® 20.0. Statistical analysis of
differences between groups was performed using the Mann-Whitney U
test. The median value and 95% confidence intervals for testosterone,
androstenedione, DHEAS and SHBG were calculated.
Results
Patient characteristics are displayed in Table 1. There were no significant
differences between groups in clinical and tumor characteristics. Also, no
differences were found between groups in PSA levels and hormonal status
(i.e. castration naïve or castration resistant prostate cancer).
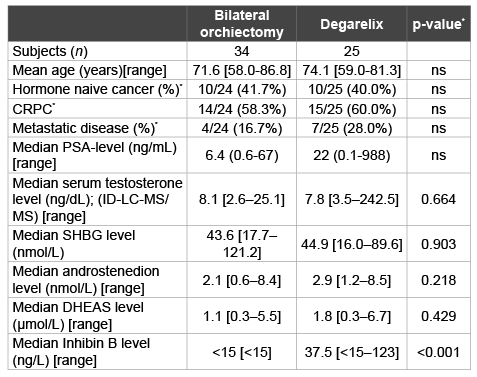
Table 1: Patient characteristics and serum hormone levels after treatment
with LHRH agonist therapy
*cancer specific characteristics in the bilateral orchiectomy group only apply
to 24 subject who underwent castration because of prostate cancer
ns: not significant; LHRH: Luteinizing Hormone-Releasing Hormone; BMI:
Body Mass Index; CRPC: Castration Resistant Prostate Cancer; ID-LC-MS/
MS: Isotope Dilution-Liquid Chromatography-Tandem Mass Spectrometry;
SHBG: Sex Hormone-Binding Globulin; DHEAS: Dehydroepiandrosterone Sulfate.
All evaluated patients had serum testosterone levels less than 50 ng/dl,
and 31 (91%) and 23 (96%) had levels less than 20 ng/dL in the surgical
castration and degarelix group respectively. Serum castrate levels of
testosterone levels were not statistically significant between those treated
with degarelix and those surgically castrated (Figure 1).
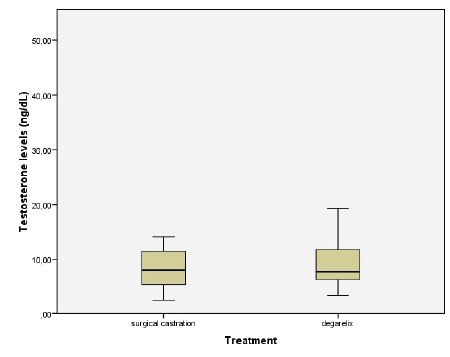
Figure 1: Box plot showing serum testosterone levels in patients after
orchiectomy and after LHRH antagonist therapy (degarelix) using IDLC-MS/MS.
Upper and lower quartiles are represented by rectangle and
maximum and minimum observed values are represented by whiskers
(outlier not shown). Median value for surgical castration was 8.1 ng/dL
and median value for degarelix therapy was 7.8 ng/dL (p=0.664)
There were no significant differences between the groups in levels of
SHBG, DHEAS and androstenedione. Patients who underwent surgical
castration had inhibin B levels below the limit of quantification, which is
significantly lower compared to the levels of inhibin B in patients treated
with degarelix.
Discussion
Bilateral orchidectomy, LHRH agonist and LHRH antagonist therapy
aim at lowering serum testosterone to a castrate level. With this, prostate
cancer growth and progression cease, signs and symptoms of advanced
or disseminated disease diminish and the lives of prostate cancer patients
may be extended. Bilateral orchiectomy achieves these goals by surgical
removal of the testes which are the primary testosterone producing organs.
LHRH antagonists primarily have their action by binding LHRH receptors
(LHRH-R) in the pituitary gland, thereby blocking the downstream
sequelae of hormone production in the hypothalamo-pitituary-gonadal
axis. Eventually, this leads to cessation of testosterone production by the
Leydig cells in the testes.
In the present study, we measured testosterone levels in men on ADT
after bilateral orchiectomy or LHRH antagonist therapy. Our data showed
that all men in the surgical castration group and all but one man in the
degarelix group achieved castrate levels of testosterone (i.e. below 50 ng/
dL). No significant difference with respect to serum testosterone level was
found between surgically castrated men and men on LHRH antagonist
therapy. This indicates that in the end, treatment with degarelix might
be as effective as surgical castration in achieving a castration level of
serum testosterone. Data on the time to achieve castration level of serum
testosterone could not be retrieved from this study. Also, no difference
was found between the two groups in levels of SHBG, androstenedion and
DHEAS. Serum levels of inhibin B were below the limit of quantification in
the surgical castration group, which implicates that the surgical castration
was complete and remaining presence of Leydig cells is unlikely.
In an earlier study from our group, it was shown that men on LHRH
agonist therapy have significant lower concentrations of serum testosterone
than men after surgical castration [3]. It might well be extrapolated
that men on LHRH agonist therapy have lower serum castrate levels of
testosterone than those on LHRH antagonist therapy.
In literature, there is some evidence that patients with advanced prostate
cancer have improved disease control with degarelix versus LHRH agonists
and that PSA progression free survival and overall survival increase.
Urinary tract events and joint and musculoskeletal events decrease with
degarelix compared with LHRH agonists [6]. Primary endpoints of these
trials were change in testosterone level, change in International Prostate
Symptom Score or prostate volume instead of survival. Also, these studies
have a short follow-up of only one year, while the median survival of
patients with newly diagnosed metastases of prostate cancer is 42 months,
which makes it difficult to draw conclusions about survival [1]. The results
of our current study suggest that these effects, if present, might not be
explained by differences in testosterone levels or by the suppression of
testosterone levels.
The differences in disease-related outcome in patients with advanced
disease treated with LHRH antagonists and LHRH agonists may be
explained by the distinct modes of action of both treatments. Most
evident, LHRH agonists stimulate the LHRH-R and LHRH antagonist
block the LHRH-R. Besides presence in the pituitary gland, the LHRH-R
is relatively highly expressed in in (benign) basal epithelial cells as well
in luminal cells of the prostate but not in the prostate stroma cells [16].
The expression is also high in breast, kidney, thymus and in lymphocytes
[16,17]. The LHRH-R can also be found in lower concentrations in the
hippocampus, the olfactory system, cerebral cortex, cerebellum, heart,
adrenal glands and the bladder [18].
In prostate cancer, the LHRH-R has been identified and the LHRH-R
expression persists despite prolonged exposure to LHRH agonists. These
receptors were also moderately to highly express in lymph node metastases
of prostate cancer [19]. Also, LHRH-Rs are expressed with high prevalence
in hormone naïve prostate cancer cells as well as in castration resistant
prostate cancer cells [19]. Other studies have shown that prostate cancer
cells have a higher expression of LHRH-Rs compared to normal prostatic
tissue [17]. The exact downstream sequelae of the stimulation of the
LHRH-R are not completely understood, but both for LHRH antagonists
and LHRH agonists it has been described in in vitro studies that they
exert a direct antiproliferative effect on human prostate cancer cells [20-
22]. It has even been suggested that the presence of LHRH-Rs in prostate
cancer leads to better clinical status and outcome of the disease [23]. These
findings imply that there could be an effect of LHRH-R targeted therapy
on prostate cancer besides the castrating effect.
Patients who underwent surgical castration had lower inhibin B levels
compared to the levels of inhibin B in patients treated with degarelix. In
an in vivo model, it was shown that inhibin suppresses prostate cancer
growth rate by almost 3-fold [24]. The role of inhibin in prostate cancer
pathogenesis and its effect on the course of the disease remain to be
clarified, but inhibin may act as a tumor suppressor in prostate cancer [25].
Besides suppression of luteinizing hormone (LH), LHRH agonists and
LHRH antagonists also suppress follicle stimulating hormone (FSH) levels
[5]. The FSH receptor is expressed in normal human prostatic tissue and in
benign prostatic hyperplasia. Interestingly, it has been shown that the FSH
receptor is expressed more intensely in prostate cancer tissue, particularly
in metastatic disease [26,27]. In tumor blood vessels, FSH receptors are
present whereas FSH receptor expression was not found in the blood
vessels of non-malignant tissues. Suppression of the levels of FSH may
thus be associated with tumor growth and tumor cell proliferation [28].
Indeed, in an in vitro model, FSH was found to increase proliferation
in the human castration resistant prostate cancer cell lines PC3 and
Du145 [29]. Different studies showed that LHRH antagonists suppress
FSH levels more profoundly than LHRH agonist [5,30]. Klotz et al. [5]
showed that FSH concentrations decreased with 89% after administration
of degarelix compared to 54.8% patients receiving leuprolide. A more
robust suppression of the FSH mediated proliferative pathway by LHRH
antagonists as compared to LHRH agonists might potentially be an
alternative mechanism by which LHRH antagonists interfere in the tumor
cell biology, thereby improving disease outcome. However, the exact
molecular mechanisms and the clinical relevance of more robust FSH
suppression by LHRH antagonists have not been fully elucidated.
There is evidence of a possible link between the LHRH-R and the
epidermal growth factor pathway (EGF). EGF is a growth factor which
is known to stimulate cell growth, proliferation and differentiation by
binding the epidermal growth factor receptor (EGFR). This binding
initiates a variety of biochemical changes in the cell (increased glycolysis
and protein synthesis amongst other things) which ultimately leads
to increased DNA synthesis and cell proliferation. Over expression of
the EGFR is associated with disease progression and poor prognosis in
prostate cancer and it has also been linked to the transition of prostate
cancer to castration resistant prostate cancer [31-33]. In other studies, it
was shown that therapy targeting the EGFR leads to inhibition of human
prostate cancer growth, possibly due to anti-angiogenic activity [34,36].
In an in vivo model, it was shown that treatment with a LHRH antagonist
decreased the level and mRNA expression of EGFR in prostate cancer
[36]. Therefore, LHRH antagonist therapy could also decelerate prostate
cancer progression through the EGFR pathway.
As mentioned before, in a large, pooled patient population comparing
degarelix with LHRH agonists, patients on degarelix had a lower risk
of death after adjusting for prognostic factors [6]. As the number of
prostate cancer deaths in this study was relatively small, differences in
disease outcome might probably be explained by a lower incidence of
cardiovascular events in the degarelix group [11]. Patients with preexisting
cardiovascular disease who were treated with degarelix had a lower risk of
experiencing a cardiovascular event (or even death) compared to patients
receiving LHRH agonist treatment with an absolute risk reduction of
8,2% in the first year of treatment [11]. Mechanisms other than the mere
suppression of serum testosterone might well be responsible for this
difference in disease outcome between LHRH antagonists and LHRH
agonists. This is particularly as the LHRH-R is expressed in the human
heart [37]. Treatment with LHRH agonists causes the lean body mass
to fall 3% with a rise in fat mass of 10% causing a 2% increase in body
weight. This change in body composition could probably alter the risk of
cardiovascular events as is the observed rise in triglycerides level (26%)
total cholesterol level (approximately 10%), and the lower body insulin
sensitivity [38]. Though, the stimulation of these LHRH-R by LHRH
agonists or otherwise, the blockage of this receptor by LHRH antagonists
has yet unknown effects on heart condition, cardiac vascularity, and the
occurrence of atherovascular disease.
Prostate cancer is considered to be a form of cancer which is highly
heterogeneic, which provides a challenging problem for clinical disease
management. Improved and detailed understanding of all genetic
alterations and variations in prostate cancer might also lead to better
understanding of clinical effects of different forms of androgen deprivation
therapy [39]. One could hypothesize that due to tumor heterogeneity,
different pathways other than the ones including androgens could
determine disease outcome. This would correspond with the findings of
this current study that the possible difference in outcome between patients
treated with LHRH agonists and LHRH antagonists cannot be explained
by a difference in serum testosterone concentrations only.
Conclusion
By using a state-of-the-art method of determination, serum testosterone
concentrations are equally reduced by treatment with a LHRH antagonist
compared to surgical castration. As there are suggestions that disease
outcome of men treated by LHRH antagonists improves as compared
to other forms of ADT such as LHRH agonists, our study showed that
mere suppression of serum testosterone level does not seems to be the
biochemical explanation for this difference. LHRH antagonists might
interfere in other hormonal and molecular pathways or otherwise directly
suppress the downstream sequelae of ligand to LHRH-R binding.
