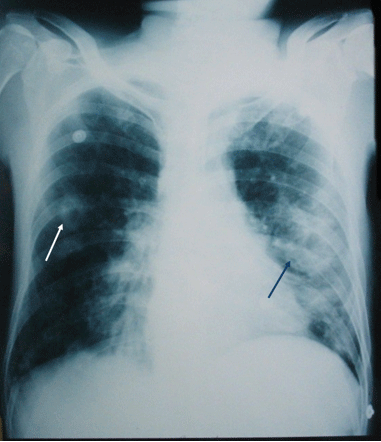
Figure 1: Initial chest X-ray (posteroanterior view): bilateral, multiple, and excavated pulmonary nodules and masses.

Salem Bouomrani1,2* Salsabil Dabboussi3,4 Nesrine Belgacem1 Safa Trabelsi1
Department of Internal Medicine, Muhimbili University of Health and Allied Sciences, Tanzania*Corresponding author: Salem Bouomrani, Department of Internal medicine, Military Hospital of Gabes, Gabes, Tunisia, Tel: +00216 98977555; E-mail: salembouomrani@yahoo.fr
Introduction: Granulomatosis with Polyangiitis (GPA), formerly known as Wegener’s disease is an autoimmune systemic necrotizing vasculitis. The mechanisms and triggers of this disease are not always well understood. We report an unusual and rapidly fatal observation of post traumatic fulminant GPA.
Case report: A 38-year-old Tunisian man, with no medical history, was initially hospitalized for hemothorax secondary to multiple costal fractures following thoracic trauma. The evolution was marked by relapsing bilateral pneumonia well controlled under adapted antibiotherapy. One month later, he reported a rapidly progressive chest pain and dyspnea. Chest X-ray and thoracic computed tomography revealed bilateral, disseminated, and excavated lung nodules and masses of variable size. Biology tests showed a marked inflammatory syndrome. The hydatid, aspergillary and HIV serologies as well as the tuberculosis screening test were negative. The evolution was fulminant with aggravation of the respiratory signs, extension of the pulmonary radiological lesions, and the installation of a rapidly progressive organic renal failure. Despite his admission to the intensive care unit and adapted resuscitation, the death occurred on the third day due to multiple organ failure syndromes.
The search for anti-neutrophil cytoplasmic antibodies was positive and autopsy exam confirmed the diagnosis of diffuse pulmonary, renal and digestive necrotizing vasculitis.
Conclusions: Our observation is characterized by its occurrence following thoracic trauma. To our knowledge, only one case of post-traumatic GPA has been reported previously.
Granulomatosis with polyangiitis; GPA; Wegener’s granulomatosis; Trauma; Post-traumatic; Vasculitis
Granulomatosis with Polyangiitis (GPA), formerly known as Wegener’s granulomatosis, is a necrotizing vasculitis of small vessels, associating diffuse inflammation of the vascular wall with peri-and extra vascular granulomatosis [1-3].
In its complete/systemic form, GPA associates ENT, pulmonary and renal involvement with classically a necrotizing glomerulonephritis representing the major prognostic factor of this disease [1,4,5]. Any other organ (kidneys, peripheral nerves, eyes, skin, joints, abdomen) can be affected by this angiitis [1,4,5].
Classically, the absence of renal involvement at the time of diagnosis defines the localized/limited forms of GPA, characterized by a better prognosis [6].
The pathogenesis of GPA is not yet fully known [2,3], although many of the mechanisms involved are now better understood. The main actors are the Cytoplasmic ANCA (C-ANCAs) auto antibodies targeting Proteinase 3 (PR3), a proteolytic enzyme contained in the cytoplasmic granules of neutrophils and monocytes [2,7]. Certain infectious and/or toxic factors may act as pathogenic factors precipitating this vasculitis in a genetically predisposed subject [3]. Trauma was only exceptionally reported as a plausible triggering factor of this systemic angiitis [8]. We are reporting an original observation of fulminant and fatal GPA precipitated by chest trauma.
A 38-year-old Tunisian man, with no pathological medical history, was initially hospitalized for thoracic trauma with multiple costal fractures complicated by haemothorax. The initial chest X-rays with right and left profiles, the costal grill, and the computed tomography showed fractures of the 3rd, 4th and 5th right anterior rib arches with minimal right hemotharax. No parenchymal lung lesions were noted.
The evolution was marked by bilateral and recurrent pneumonia caused by Staphylococcus aureus and then by Pseudomonas aeruginosa well controlled by an adapted antibiotherapy. He was discharged from the hospital after a month without any abnormalities; his last chest X-ray before discharge was particularly normal.
One week after discharge, the patient reported a rapidly progressive chest pain and exertional dyspnea for two days. There was no reported fever or associated expectoration. The somatic examination noted anon febrile and dyspneic (respiratory rate at 22 cycles/min and spontaneous oxygen saturation at 82%) patient with a stable hemodynamic and neurological status. The examination has not shown skin lesions, palpable peripheral lymphadenopathy, visceromegaly, and palpable masses.
The electrocardiogram revealed a simple regular tachycardia at 110/min. The biology tests showed a marked inflammatory syndrome with an erythrocyte sedimentation rate at 130 mm/H1, a C reactive protein at 48 mg/l, and a moderate leukocytosis at 12500/mm3 with 75% neutrophils.
Other basic biological tests were within normal limits (platelets, hemoglobin, creatinine, glycaemia, serum calcium, transaminases, muscle enzymes, total cholesterol, triglycerides, serum protein electrophoresis, and urine analysis). Chest X-ray and thoracic CT showed multiple, bilateral and excavated lung nodules and masses of varying size (Figures 1 and 2).
Figure 1: Initial chest X-ray (posteroanterior view): bilateral, multiple, and excavated pulmonary nodules and masses.
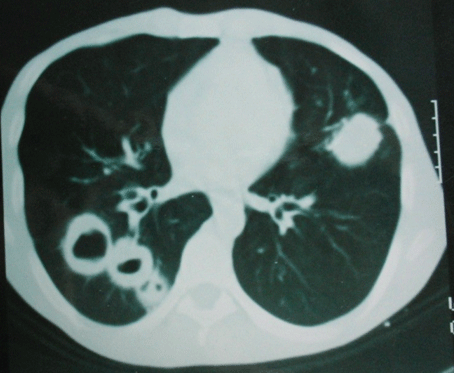
Figure 2: Initial thoracic CT in axial section with contrast: multiple excavated and thick walled pulmonary nodules.
The hydatid, aspergillary and HIV serologies as well as the tuberculosis assessment were negative. The blood cultures and the transthoracic echocardiography were also without abnormalities. Considering the negativity of these tests and the history of recent thoracic trauma, the diagnosis of multiple bilateral traumatic pulmonary pseuodocysts was suspected.
The evolution was fulminant with rapid aggravation of the respiratory signs, extension of the pulmonary radiological lesions at the third day (Figures 3 and 4), and installation of a rapidly progressive organic renal failure with creatinine at 340 µmol/l the third day and 790 μmol/l the fourth day. Despite his admission to the intensive care unit and adapted reanimation, the death occurred on the sixth day because of multiple organ failure.
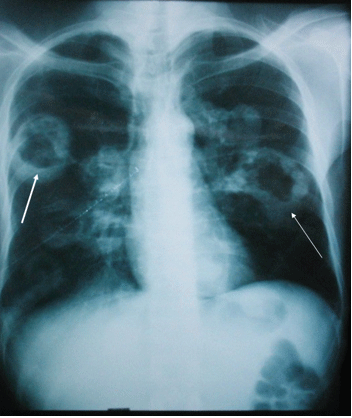
Figure 3: Chest X-ray after 3 days: extension and multiplication of bilateral and excavated pulmonary nodules and masses.
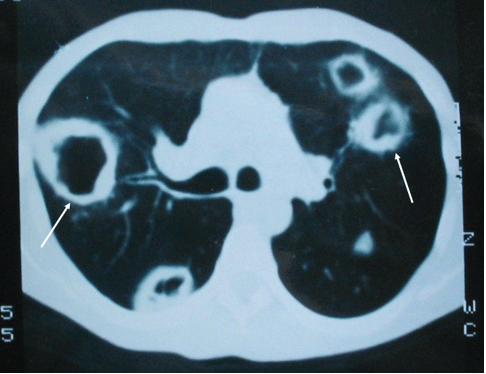
Figure 4: Thoracic CT in axial section with contrast after 3 days: multiple bilateral, excavated, and thick-walled pulmonary nodules.
Screening for anti-neutrophil cytoplasmic antibodies with EnzymeLinked Immunosorbent Assay (ELISA) was positive and at a very high titer (1/640), and a diffuse cytoplasmic staining pattern was noted in indirect immunofluorescence.
An autopsy was indicated, and histological examination confirmed the diagnosis of diffuse pulmonary, renal and digestive necrotizing vasculitis with images of granulomatous and necrotizing vasculitis of small caliber vessels (mainly arterial and capillary) with a large polymorphic inflammatory infiltrate in the kidneys, lungs, myocardium, stomach and intestines. Thus the diagnosis of fulminant and fatal GPA occurring after a thoracic trauma was retained.
GPA is a relatively rare but potentially fatal systemic vasculitis in the absence of an urgent and adequate treatment. Its prevalence is estimated at about 5 per 100,000 inhabitants, and appears more frequent among Caucasian people and in Nordic countries [1-3]. Its 10-year survival rate is estimated at 40% if there’s a renal impairment and 60-70% for forms without renal involvement [3,9].
The prognosis of this vasculitis was clearly transformed by the new therapeutic protocols combining systemic corticosteroids and immune suppressants, particularly cyclophosphamide, and more recently the biotherapy [10,11]. In fact, the mortality of this ANCA-associated vascualitis has significantly decreased with the wide use of these therapeutic protocols: in the Wallace ZS, et al. [12] series, comparing two GPA chronological cohorts (1992-2002 and 2003-2013), mortality was reduced in the late cohort, thanks to new therapies (35.7 vs. 72.0 cases per 1000 person-years).
As a result, rapid diagnosis and therapeutic intervention without delay are the only guarantors to improve the prognosis of this disease [1].
In fact, the GPA presentation can be rapidly progressive [13], fulminant [14,15], and even fatal [16,17]. Pathophysiologically, the exact mechanisms favoring the development of this vasculitis are not yet well understood [2,3]. This angiitis appears to be multifactorial, involving immune dysfunction as the main etiological factor (GPA is considered as an autoimmune vasculitis, associated with ANCA); an infectious, toxic or environmental precipitating factor, and a particular genetic predisposing terrain [1-3].
Trauma, as a potential precipitating factor, has been reported only once in the world literature: it is a localized form of ocular GPA precipitated by facial trauma with an orbital floor fracture, in a man of 31 years old with no medical history [8]. The trauma appears to act as a stimulus for the inflammatory reaction, triggering and precipitating the course of this vasculitis [8].
GPA is a relatively rare systemic necrotizing vasculitis that may have atypical, unusual, sometimes rapidly progressive, fulminant, and even fatal presentations. This imposes a better knowledge of this disease with its various possible clinical facets, in order to allow rapid diagnosis and appropriate management without delay, given the spontaneously fatal nature of this disease.
Our observation is characterized by its occurrence following thoracic trauma. To our knowledge, the possible role of trauma in the development of GPA has only been reported once.
None
Download Provisional PDF Here
Article Type: CASE REPORT
Citation: Bouomrani S, Dabboussi S, Belgacem N, Trabelsi S (2018) Post-Traumatic Fatal Granulomatosis with Polyangiitis. Clin Res Open Access 4(4): dx.doi.org/10.16966/2469-6714.142
Copyright: © 2018 Bouomrani S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access