Abstract
The term “hypereosinophilic syndrome” refers to a rare group of disorders characterized by a persistent, marked proliferation of eosinophils
with end-organ involvement. Chronic eosinophilic leukemia is a myeloproliferative variant of hypereosinophilic syndrome characterized by clonal
eosinophilia, which can result in hematologic, cardiac, or pulmonary end-organ damage, among others. We present a case of chronic eosinophilic
leukemia seen at our institution and discuss an approach to making the diagnosis of hypereosinophilic syndrome in general, and chronic
eosinophilic leukemia in particular. We also explore treatment options in the management of hypereosinophilic syndrome/chronic eosinophilic
leukemia, including novel agents like Alemtuzumab and Mepolizumab.
Keywords
Chronic eosinophilic leukemia; Hypereosinophilic syndrome; Imatinib; FIP1L1-PDGFRA
Introduction
Eosinophils are non-dividing, end stage cells that differentiate from the
hematopoietic stem cell in the bone marrow. They migrate in the blood
transiently and are predominantly tissue-dwelling cells [1]. Eosinophils
play a pivotal role in the body’s response to parasitic infections and some
bacterial infections, and are also important in the etiopathogenesis of
atopy and allergy reactions. Although eosinophilopoiesis and egress from
the bone marrow is regulated by T cell-mediated cytokines depending
on presence of allergens or infections [1], production is occasionally not
controlled by these mechanisms and a hypereosinophilic state may result.
The term “hypereosinophilic syndrome”(HES) has been described
to explain the finding of persistent eosinophilia of 1.5 × 109
/L or higher
(≥ 1500 eosinophils/mm3
) lasting greater than 6 months, with evidence
of organ involvement, and in the absence of other known causes of
eosinophilia such as parasitic infection or allergic reaction [2]. Six
clinical types of HES were described at a 2005 international consensus
workshop on the treatment of HES: (i) myeloproliferative variant HES,
(ii) lymphocytic variant HES, (iii) familial HES, (iv) overlap HES, (v)
associated HES and (vi) idiopathic HES [2]. Chronic eosinophilic
leukemia (CEL) falls within the myeloproliferative variant of HES. It is
characterized by clonal eosinophilia and can be differentiated from the
broad category of HES by the presence of increased peripheral blood and
marrow blasts or by the demonstration of a clonal cytogenetic abnormality
or a hallmark tyrosine kinase activating mutation in the myeloid lineage
[3]. CEL discriminately affects men, with a male-to-female ratio estimated
at 9:1 [3]. Peak incidence occurs between ages 20-50, although some cases
have been reported in infants and children [3]. The clinical features seen
in CEL include hepatomegaly, splenomegaly, anemia, thrombocytopenia,
and bone marrow dysplasia or fibrosis. Cardiac involvement may lead
to endomyocardial fibrosis and valvular insufficiency, and pulmonary
involvement can cause fibrosis, effusions, emboli, and ground glass
attenuation, among others. Patients can have elevated cobalamin and
tryptase levels as well as increased levels of atypical mast cells [2,3].
We present a case of CEL seen at our institution and discuss an
approach to the diagnosis and management of CEL in particular, and HES
in general.
Case Report
The patient is a 54-year-old African-American male with history
of asthma, (requiring intubation in the past for an asthma flare), who
presented with sudden onset pleuritic chest pain and dyspnea. Chest
x-ray was normal and cardiac work-up was negative. He was diagnosed
with acute asthma exacerbation, admitted, and was treated with IV methyl
prednisolone, albuterol and ipratropium nebulizers with improvement in
his symptoms. He was found to have significant eosinophilia on admission,
which had been longstanding (for over 3 years) on review of his medical
record. Laboratories showed white blood cell count of 24.6 × 109
/L
with a differential of 9% neutrophils, 1% bands, 19% lymphocytes, 6%
monocytes, and 45% eosinophils. Hemoglobin was 10.9 g/dL, hematocrit
33.8 %, platelets 155 × 109
/L, and MCV 85.8. Hematology consultation
was therefore sought for marked eosinophilia.
On evaluation by the hematology team, he denied fevers, chills, night
sweats, weight loss, abdominal pain, nausea, vomiting or diarrhea. He
denied recent travel abroad, camping, or exposure to unsanitary water or
food. He also denied easy bruising or bleeding, allergic reactions, or history
of allergy to medication. His home medications were albuterol and Advair
inhalers. He endorsed a 10-pack-year history of smoking but had quit 30
years prior. He, however, did report occasional cocaine use. His family
history is significant for asthma in his mother who died of asthma-related
complications. His physical examination revealed diffuse inspiratory and
expiratory wheezing in all lung fields and moderate splenomegaly, but was
otherwise unremarkable. He had no palpable lymph nodes.
A review of his medical record revealed that he had been evaluated
for hypereosinophilia on a prior admission for asthma exacerbation, 3
years before his current presentation. A bone marrow biopsy at that time
had shown markedly hypercellular marrow with marked myeloid and
eosinophilic hyperplasia, and florescent in-situ hybridization (FISH) had
revealed the presence of the FIP1L1-PDGFRA (Fip1-like-1 fused with
platelet derived growth factor receptor alpha) mutation consistent with
chronic eosinophilic leukemia. Unfortunately, he was lost to follow up
until this admission.
To guide management, we reviewed the patient’s peripheral blood
smear and he underwent a repeat bone marrow biopsy which showed
a markedly hypercellular marrow (95% cellularity) with predominant
eosinophilia (60% of total marrow cellularity) (Figure 1). Flow cytometry
showed myeloid predominance with increased CD52+ eosino forms;
and florescent in-situ hybridization (FISH) was positive for the FIP1L1-
PDGFRA mutation, consistent with chronic eosinophilic leukemia.
Cytogenetic analysis revealed normal karyotype. We also obtained
echocardiography, CT scan of the chest and pulmonary function
tests, looking for other evidence of organ involvement but these were
unremarkable. His only evidence of organ involvement was the bone
marrow findings, mild anemia and splenomegaly.
Given that patients with FIP1L1-PDGFRA-mutated CEL have virtually
universal response to Imatinib, we encouraged the patient repeatedly on
multiple occasions to begin treatment with Imatinib. Unfortunately, he
declined treatment. It has been about a year since his last comprehensive
hematology review. He has been readmitted twice subsequently in the
interim period for asthma flares for which he received routine treatment
and was discharged. We reiterated the need to receive treatment for CEL
on these occasions but he remains yet unwilling. Interestingly, he showed
no overt signs of deterioration in his clinical or performance status from
his initial presentation.
Discussion
The term “hypereosinophilic syndrome” (HES) was first coined in
1975 by Chusid et al. to describe patients with profound eosinophilia
of an unclear cause [3]. To meet criteria for this diagnosis, patients had
to demonstrate: (i) Persistent eosinophilia of 1.5 × 109
/L (1500/mm3
) or
higher for a period greater 6 months; (ii) absence of other known causes
of eosinophilia; and (iii) signs and symptoms of end organ involvement.
The initial criteria established in 1975 are still used in making the
diagnosis today. A patient suspected of having HES owing to prolonged
profound eosinophilia should first undergo rigorous evaluation to
rule out secondary causes of eosinophilia including parasitic, bacterial,
fungal or viral infection; allergic and drug hypersensitivity reactions;
neoplasms like leukemias, lymphomas, or solid organ adenocarcinomas;
and autoimmune disorders or connective tissue disease [4]. Other causes
such as hypoadrenalism, radiation exposure, cholesterol embolization and
IL-2 therapy should also be ruled out [4]. Failure to identify a secondary
cause for the eosinophilia should then lead to a comprehensive work-up
to identify end-organ damage from HES and a possible clonal population
of eosinophils, as is the case with myeloproliferative variant HES and CEL.
Work-up should include routine blood studies such as complete blood
count with differential and chemistries, serum troponin, echocardiogram,
computed tomography scans of the chest/abdomen/pelvis, and pulmonary
function tests to establish end-organ involvement. A biopsy of affected
tissues can also be undertaken if feasible [4]. A review of the peripheral
smear and screening of the peripheral blood for the FIP1L1-PDGFRA
(F/P) mutation by FISH or reverse transcription polymerase chain reaction
(RT-PCR) is crucial in identifying clonal eosinophilia, as is the case in CEL
[2-5]. If screening for the F/P mutation is negative, bone marrow biopsy
and cytogenetic analysis should be undertaken to look for other evidence
of clonal eosinophilia such as 5q33 and 4q12 translocations, which suggest
PDGFRB (platelet derived growth factor receptor beta) and PDGFRArearranged
clonal eosinophilia respectively [5]. These translocations
portend favorable response to Imatinib [5]. Analysis may, however, reveal
8p11.2 translocation, which suggests FGFR1 (fibroblast growth factor
receptor 1)-rearranged clonal eosinophilia, associated with aggressive
myeloid malignancies that are refractory to current drug therapy [5].
Bone marrow evaluation is also useful because it is helpful in excluding
other well-defined myeloid malignancies, which can be secondary
causes of eosinophilia. Failure to identify a clonal population on bone
marrow evaluation should prompt investigation for an aberrant or clonal
lymphocyte population with T cell receptor (TCR) gene rearrangement
studies and peripheral blood lymphocyte phenotyping [5]. Identification
of an aberrant/clonal lymphocyte population in this setting makes the
diagnosis of lymphocytic variant HES, whereas the failure to identify such
a population suggests a diagnosis of idiopathic HES [5].
Hypereosinophilic syndromes were historically treated with
corticosteroids primarily, with hydroxyurea and interferon-alpha reserved
as second line therapies. However, with reports of improved survival with
Imatinib in chronic myelogenous leukemia (CML) in the early 2000s,
Physicians started to use it in treating patients with HES/CEL based on
the hypothesis that both CML and HES/CEL share a common pathogenic
mechanism [3]. The first report of Imatinib use in HES was in 2001, in a
patient with HES refractory to corticosteroids, hydroxyurea, and interferon
alpha. He was given Imatinib and achieved complete hematologic response
after taking Imatinib 100 mg daily for only 4 days [3]. A subsequent paper
documented response to Imatinib 100 mg daily in 4 of 5 patients who
were treated with this regimen [6]. Yet another study showed Imatinib
responsiveness despite high serum IL-5 levels, demonstrating that the
level of eosinophil-associated cytokine production was not necessarily
predictive of Imatinib responsiveness in HES [3,7]. Several other patients
with HES/CEL also showed good response to Imatinib, and a landmark
study by Cools et al later identified that the molecular basis for response
to Imatinib in HES was the inhibition of a novel fusion tyrosine kinase:
FIP1L1-PDGFRA (F/P) [3,8].
Patients with F/P positive CEL or PDGFR-associated CEL should be
treated with Imatinib (100-400 mg by mouth daily) given that response
to Imatinib in these patients is almost universal, with patients achieving
complete hematologic and molecular remission within days to weeks [2].
Maintenance therapy with daily Imatinib and surveillance with FISH or
RT-PCR checking for the reappearance of the FIP1L1-PDGRFA fusion
transcript (molecular relapse) every 3 to 6 months is recommended
[9]. PDGFR-negative HES/CEL is not as responsive to Imatinib with
reported response rates ranging from 9-60 % [2]. These patients are
treated traditionally with corticosteroids. If refractory to corticosteroids,
however, they are treated with Imatinib, but typically require higher doses
and longer duration of therapy to achieve remission [2,10]. If refractory to
Imatinib as well, other possible options for therapy include hydroxyurea,
interferon alpha, second-and third-generation tyrosine kinase inhibitors,
and allogeneic stem cell transplantation [2]. Interferon alpha provided
a good response in a patient who was treated for coexisting CEL and
Hepatitis C infection, with a significant decline in his eosinophilia and
improvement in his symptoms after beginning interferon therapy [11].
Lymphocytic variant HES (L-HES) is initially treated with corticosteroids.
Interferon alpha is the preferred second line therapy given its effect on both
eosinophils and T cells [2]. In the case of idiopathic HES, corticosteroids
are also first line therapy, with hydroxyurea and interferon alpha reserved
as possible second line agents [2-5].
Novel agents in the management of HES and/or CEL have been
investigated and show great promise. Two of these are Alemtuzumab
and Mepolizumab, both of which are humanized monoclonal antibodies.
Alemtuzumab is an anti-CD52 antibody, which was investigated as a
potential effective therapy in HES due to the inherent expression of CD52
on eosinophils. A study at the MD Anderson Cancer Center in Houston,
Texas, USA showed remarkable response rates and durable complete
hematologic remission, especially with maintenance therapy [12].
Interestingly, some patients were able to achieve up to a third remission
after repeat induction therapy with Alemtuzumab upon relapse [12]. In
the case of Mepolizumab, it was postulated to be a potential therapeutic
agent in HES since it binds with high affinity to IL-5, preventing it from
interacting with its receptor on eosinophils. IL-5 is known to play a
significant role in eosinophil maturation, differentiation, mobilization,
activation, and survival [13]. A study by Rothenberg et al to evaluate the
effects of mepolizumab on corticosteroid sparing and the maintenance of
clinical stability in patients with HES treated with corticosteroids showed
that it is effective and can result in corticosteroid-sparing for patients
with FIP1L1-PDGFRA negative hypereosinophilic syndrome [13]. At the
moment Alemtuzumab and Mepolizumab are only available on clinical
protocols for refractory HES or on compassionate use basis and are not
yet mainstream therapy for HES and/or CEL.
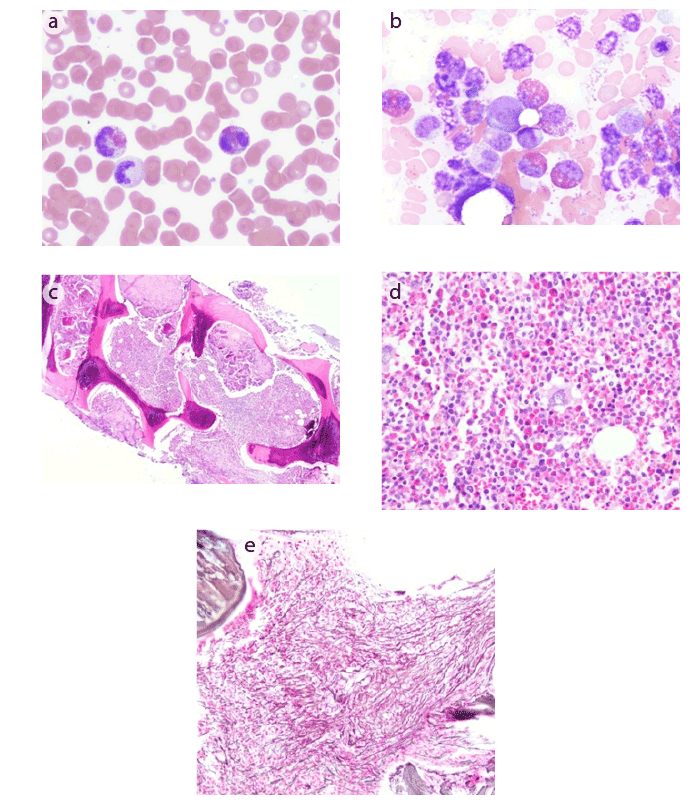
Figure 1: (a) Peripheral blood showing marked eosinophilia, with some eosinophils showing degranulation
(b) Bone marrow aspirate showing a marked increase in eosinophils and their precursors
(c) Bone marrow core biopsy showing hypercellularity with a greater than 90% cell to fat ratio
(d) Bone marrow core biopsy demonstrating sheets of eosinophils and their precursors
(e) Reticulin stain of bone marrow core biopsy showing a moderate to marked increase in reticulin fibrosis
Conclusion
HES/CEL is a group of rare disorders characterized by a persistent
marked proliferation of eosinophils with end organ involvement. Despite
advances in treatment with the discovery of Imatinib in patients with
the FIP1L1-PDGFRA mutation, further research is needed for the
development of new therapies. Alemtuzumab and Mepolizumab are
two novel agents that show promise, but are yet to become mainstream
therapy for managing HES/CEL.
Declaration of Interests
The authors state no conflict of interests and have received no payment
in the preparation of this paper or in conducting the study.
Article Information
Article Type: Case Report
Citation: Ogbonna OH, Nwabudike SM, TaddesseHeath
L, Oneal P (2016) Chronic Eosinophilic
Leukemia in an African American Man. Clin Res Open Access 2(1):
doi http://dx.doi.org/10.16966/2469-6714.113
Copyright: © 2016 Ogbonna O H, et al. This is
an open-access article distributed under the terms
of the Creative Commons Attribution License,
which permits unrestricted use, distribution, and
reproduction in any medium, provided the original
author and source are credited.
Publication history:
Received date: 08 Dec 2015
Accepted date: 28
Jan 2016
Published date: 03 Feb 2016
