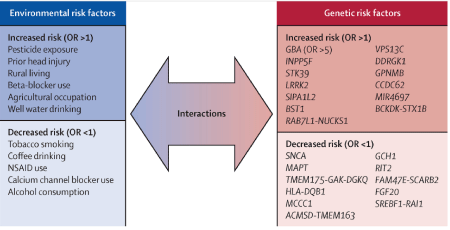
Figure 1: Factors that increase and lower the risk of PD. Taken from “Parkinson’s Disease” [10].
Laura Burckhalter Vincent S Gallicchio*
Department of Biological Sciences, College of Science, Clemson University, Clemson, USA*Corresponding author: Vincent S Gallicchio, Department of Biological Sciences, College of Science, Clemson University, Clemson, USA, E mail: vsgall@clemson.edu
Parkinson’s disease (PD) is a neurodegenerative disease characterized by loss of dopaminergic neurons in the substantia nigra. Current treatment only helps symptoms, and there is no effective treatment to delay or reverse PD. Scientists have regarded a number of genetic and environmental factors that may lead to developing PD. Lithium has been used for years in psychiatry as an effective mood stabilizer in manic depressive bipolar disorder (BD). This natural element is the most potent inhibitor of the enzyme glycogen synthetase kinase (GSK3) activity. Because of GSK3’s abundance in the body and over expression leading to PD, lithium has been postulated be used as a treatment for numerous neurodegenerative diseases and conditions where GSK3 is over expressed. Lithium has also been shown to be protective to cells against rotenone-induced PD. In contrast cellular based therapy, specifically the use of induced pluripotential stem cells (iPSCs), also pose a promising therapeutic modality for future use in order to more fully understanding the process of PD development.
Parkinson’s disease; Lithium; Stem cell; Neurodegenerative; iPSCs
Parkinson’s disease (PD) is a neurodegenerative disease that affects the dopamine producing neurons in the substantia nigra of the brain causing tremors, postural instability, and slow movements, along with non-motors appearing as diarrhea, sleeping disorders, and difficulty swallowing [1,2]. Parkinson’s disease was first described in 1817 by James Parkinson [3]. Analyses estimate that PD affects 12-15 people per 100,000 of the US population [4] and affects about 1.8 times more men than women [5]. The estimation of PD is expected to rise from 4.1 million cases in 2005 to almost 8.7 million cases worldwide by 2030, and this drastic increase is due to a population that continues to age [6].
Lithium has been the treatment of choice for manic depressive bipolar disorder (BD) since the mid-1950’s [7]. Lithium is prescribed in the form of lithium carbonate. Just as any drug, lithium has its own therapeutic plasma level. The most common range for the maintenance of bipolar disorder is between 0.6-0.8 mmol/L plasma. This dosage can be lowered to treat depression, or it can be increased for mania. However, the levels should rarely rise above 1.0 mmol/L plasma to prevent toxicity. Physicians must regularly monitor blood plasma levels of their patients to ensure the most effective use of the treatment while preventing toxicity. Dosing may need to be manipulated depending on the age of the patient. Children and older patients usually reach therapeutic levels with smaller doses [8].
Lithium has also been shown to increase production of the bone marrow, specifically hematopoietic and mesenchymal stem cells [9]. Stem cells are known for their ability to differentiate into multiple kinds of cells. For years scientists have been finding ways to use stem cells to treat diseases in which cells of a certain type have degenerated. This includes neurodegenerative diseases that specifically attack neurons; therefore, the underlying question remains can scientists find a way to use stem cells to treat Parkinson’s disease? This review focuses on the connections between Parkinson’s disease, lithium treatment, and stem cell therapy.
Parkinson’s disease is a progressive neurodegenerative disease primarily affecting the substantia nigra by causing the death of dopaminergic neurons [7]. Aggregates of alpha-synuclein proteins in Lewy bodies are also common in the neurons that remain [10]. These neurons normally produce dopamine which functions in the direct and indirect pathways of the nigrostriatal pathway. The direct pathway acts to stimulate voluntary movements, while the indirect pathway inhibits these movements. When dopamine is unable to be created or released by degenerating neurons due to Lewy bodies, the receiving end of the pathways, the striatum, has more inhibitory motor action. This gives reasoning to the slow movements and other motor symptoms seen in PD patients [11].
The symptom most attributed to PD is tremors, but it is only one of many motor symptoms that are also accompanied by non-motor symptoms. The motor symptoms of PD vary in patients along with severity [10]. Most notably, they are muscle rigidity, postural instability, dyskinesia, slowed movement, and gait impairment. Researchers have attempted to designate categories of tremor-dominant patients and non-tremor-dominant. Still yet, a large majority of patients fall somewhere between these two extremes, showing symptoms from both categories. The tremor-dominant group has shown to progress more slowly, while the non-tremor-dominant is more progressive [10].
Non-motor symptoms of PD include sleeping disorders, drooling, and cognitive impairment [12]. These symptoms typically present themselves years before motor symptoms, sometimes by up to a decade. This is called the pro-dromal phase of PD [13]. Other symptoms during this stage include depression and constipation. Researchers have suggested that if a treatment were implemented to reverse or delay the disease, this is the stage that would most benefit the prognosis of the patient [14]. However, these symptoms can easily be attributed to old age, making detecting PD difficult until the motor symptoms present themselves. By this time, the advantageous period of the pro-dromal stage has passed. Symptoms will continue to worsen, leading to falls, speech impairment, and difficulty eating due to choking. These symptoms are particularly resistant to treatment and often the cause of institutionalization of the patient to ease the stress of the caretaker [10].
The causes of PD remain relatively unknown, but researchers have separated PD into familial and sporadic. The causes of sporadic PD are unknown, but familial PD is linked to mutated genes leading to the disease. Polymeropoulos, et al. [2] located missense mutations in SNCA on chromosome 4 that lead to PD. Various single-nucleotide mutations found on this gene lead to early onset of the disease. This gene is responsible for making alpha-synuclein and having two copies of the SNCA locus led to this protein’s over production. Having three copies increased severity of the disease. The LRRK2 gene and the VPS35 gene are added into the same category as SNCA in that they are all autosomal dominant [15]. LRRK2 stands for leucinerich repeat kinase 2. This gene controls attributes of neurons such as their morphology. It also controls the reuptake of hormones and chemicals in synapses, as well as dopaminergic receptors. Scientists have discovered that this gene plays a role in processes involving mitochondrial health and autophagy [16]. The responsibilities of this gene play a role in PD characteristics. The gene encoding for Parkin, PARKIN, is autosomal recessive, along with PINK1 and DJ-1 [17]. The genes encoding PINK1 and Parkin assist in mitochondrial upkeep of a cell. When a cell is damaged, it must be destroyed via mitophagy. PINK1 is normally brought into healthy mitochondria; however, it accumulates on the membrane of damaged ones. It then recruits Parkin, which tags the mitochondria with ubiquitin for its degradation. When the regulation of these genes is out of control, this may lead to an abnormal management of mitochondria. The build-up of alphasynuclein proteins as Lewy bodies throws off the natural process of mitophagy in neurons [18]. While genetics are the cause of familial PD, they only account for a small percentage of sporadic PD cases [16]. Environmental factors can also increase the risk of developing PD.
Another genetic factor for PD can be found in the PARK15 gene. This gene encodes for the F-box only 7 (FBXO7) protein. It is part of the F-box family; however, it does not contain other domains [19]. The FBXO7 protein is necessary to successfully recruit and phosphorylate Parkin in mitochondrial health and mitophagy [20]. PTEN-induced kinase 1 (PINK1), Parkin, and the FBXO7 protein must all bind together so that PINK1 can phosphorylate Parkin. Mutations in the ubiquitin related (UbR) domain of FBXO7 reduces the ability of the FBXO7 protein to bind to Parkin, but these mutations do not obstruct its ability to bind to PINK1 [20]. However, all three components must be bound together for Parkin to be phosphorylated and retain mitochondrial health through mitophagy. Studies have shown that an over-expression of the wild-type causes FBXO7 to aggregate mostly in the cytoplasm [21]. This study also found that exposure to dopamine and H2O2 causes the aggregates to worsen. These types of aggregates have been found in PD patients [22].
The GBA gene encodes for an enzyme found in the lysosome called glucocerebrosidase (GCase). This enzyme is responsible for degrading glucocerebroside into ceramide and glucose, and also cleaves glucosylsphingosine, but not as much [23]. This enzyme can be cleaved in two separate ways to give two different sized proteins, each of which play a different role in how it enters the endoplasmic reticulum (ER) before eventually making its way to the lysosome [24,25]. This gene and its associated mutations are more commonly known for causing Gaucher’s disease, and GBA mutations are common in AshkenaziJews [26]. By studying these people that also developed PD, scientists discovered the link between GBA and PD [27]. Around 300 mutations have been found [28] and they all have differing results on the functionality of the enzyme. Some mutations cause GCase to remain in the ER [29], which may increase the risk of synucleinopathies and PD [30,31].
There are two suggested mechanisms for how the GBA gene and its mutations contribute to higher PD risk. One is that glucosylceramide leads to an accumulation of alpha-synuclein; this in turn leads to a decrease in GCase activity [32]. This proposal is supported by studies with mice and cell models [33]. Lower GCase activity has been seen in the brain tissue [34] along with the cerebrospinal fluid [35] and peripheral blood of PD patients [36]. A second hypothesized mechanism for these mutations and their relation to higher PD risk is that alpha-synuclein accumulation leads to more ER stress and ER-associated degradation (ERAD) impairment [37]. A supporting observation is the association of PARK2 and other PD associated genes in ERAD [38,39]. However, not all of these hypotheses are supported, based on the fact that in some GBA mutations, no protein is produced at all [30], yet PD risk is still increased [40]. Therefore, this higher risk cannot be due to the aggregation of these proteins. There is a higher reasonability that more ER stress is due to aggregates of alphasynuclein and not aggregates of GCase.
Coenzyme Q2 (COQ2) is encoded by the COQ2 gene [41]. It is also known as polyprenyl transferase, and mutations in this gene have been linked to two family cases of multiple-system atropy (MSA) [42]. This is also a neurodegenerative disease that gives rise to Parkinsonism symptoms along with cerebellar ataxia, failure of the autonomic system, and corticospinal disorders [43]. For years this disease was not considered to have any genetic risks or components. However, after discovering the COQ2 connections in two separate families with MSA, scientists began to look deeper. Considering both MSA and PD are alpha-synucleinopathies, an interest in the relation between the two neurodegenerative diseases peaked. COQ2 is needed in the second step of making COQ10 [44]. COQ10 is an antioxidant, stabilizes membranes, and also transports electrons [45]. Without this enzyme, people can suffer from seizures, ataxia, and mental retardation along with other symptoms [46]. Researchers have hypothesized the role of COQ2 in neurodegenerative diseases is to disturb redox reactions in mitochondria and increasing the oxidative stress [45]. Other mutations known to have an effect in development of PD also use mechanisms related to mitochondrial function such as Parkin, PINK1, and FBXO7 [20]. Therefore, COQ2 could potentially induce PD symptoms as well.
In 2015, Funayama studied a large Japanese family in which multiple people carried mutations in CHCHD2 [47]. Another family was found to have this same mutation that also carried autosomal dominant PD [47]. In a separate family from China, two siblings had the same mutation in this gene as well as a family history of autosomal dominant PD. Both siblings showed tremors and reduced reuptake of dopamine in the brain [48]. CHCHD2 codes for a transcription factor that activates COX4I2 which is a respiratory chain protein found in mitochondria. Other studies have observed mutations in CHCHD2. Specifically, the mutation pGln126 is thought to be pathogenic for PD by leading to the truncation of proteins [49]. When studying this gene in Drosophilia, a dysfunction in locomotor activity and a loss of dopaminergic neurons can be observed [50].
Another gene that has been theorized to be linked to PD is eIF4G1. This gene encodes the protein eukaryotic translation initiation factor 4-gamma-1 (eIF4G1) [51]. This protein is needed in a complex called eukaryotic initiation factor 4F (eIF4F) to begin translation [52]. Other players in this initiation complex are PABP, EIF3, and EIF4F which has two pieces: eIF4E and eIF4A [52]. Together these components come together to create the complex that recognizes a cap structure on mRNA. The complex brings the mRNA to the ribosome subunit 40S so translation can begin. Mutations have been found that weakens the binding between eIF4G1 and eIF4E which is unable to bring the mRNA to the ribosome [53,54]. Other mutations found in PD disrupt the binding between eIF4G1 and eIF3E which is believed to create a bridge between the mRNA cap and 40S unit of the ribosome [54,55]. Both of these mutations affect how a cell responds to stress.
Incidence of PD is low below 50 years of age and increases drastically after. However, a decrease in the diagnosis of PD with increasing age has been noticed, and it is thought to be due to the increasing amount of dementia [56]. PD is ruled out of possible diagnoses if dementia is present when these motor symptoms begin [56,57]. Although a rather small difference, men are often at a higher risk than women. A study by a health organization in the US determined Hispanics to have the highest rate of PD, followed by Dairy consumption has been shown to have a correlation with increase in PD [58-60]. This connection was stronger in men than in women. Scientists have not determined the reason for this connection to increase in PD risk, but it is not due to vitamin D or calcium [58]. In similar instances, high milk consumption was associated with a decrease in the density of neurons in the substantia nigra of people that do not smoke [61]. A contaminant in milk could be due to this association, but scientists lean more to the ability of milk to lower urate concentrations, which is shown to be a preventative factor for PD [56,62].
Pesticide exposure has also been correlated with an increase in PD [63]. Specifically, people that maintain jobs where they are consistently exposed to these chemicals show the increase of PD. Organochlorine pesticides were not shown to be associated with PD, so other classes must be responsible [64]. The specific chemicals in pesticides correlating with PD have yet to be determined. However, rotenone, a pesticide and insecticide has been linked to PD [65]. This substance is extremely lipophilic which makes it able to cross membranes including the blood-brain barrier [65]. Rotenone inhibits complex I which decreases ATP and allows electrons to escape that can become reactive oxygen species, increasing oxidative stress on cells [65,66]. Models have been created using rotenone to induce PD in animals showing the motor and non-motor symptoms, which has been difficult to obtain prior to rotenone. Using these models, scientists may be able to better understand the development of PD and preventable measures.
Another way to induce PD in animal models is through 1-methyl4-phenylpyridinium (MPP+) induced PD. MPP+ induced PD was discovered in an interesting way. A patient came into the hospital displaying PD symptoms that occurred overnight along with being unresponsive while appearing alert [67]. After some research, doctors discovered the patient had taken synthetic heroine consisting of almost pure 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Upon further research, scientists discovered that MPTP is not toxic, but when it reaches the brain it is converted to 1-methyl-4- phenylpyridinium (MPP+) by monoamine oxidase [68,69]. This happens mainly in astrocytes which release it [70]. MPP+ is an excellent substrate for dopaminergic neuron receptors, and in these neurons MPP+ concentrates extensively in mitochondria and blocks complex 1 [71].
Significant research has been done on the correlation between increased risk of PD and melanoma [72]. A large study of PD patients also showed a correlation with melanoma [73]. People with melanoma are at a 44% increased risk of PD [73]. The reason is still unknown. Traumatic brain injury has been associated with an increased risk of PD as well [74-77]. Injury of the brain leads to chronic inflammation, a decrease in the functionality of the blood brain barrier, damage to mitochondria, and excess alpha-synuclein [56]. All of these could lead to PD. However, an interesting correlation was discovered about head injury and PD. The risk of PD is highest soon after the trauma and decreases close to a normal risk level after 10 years [74]. Scientists believe this is reverse causation, and the head injury is due to complications of PD such as gait impairment and falls [56]. This, in turn, would lead to more diagnoses of PD soon after hospitalization for head trauma. Diabetes has a complicated correlation with PD. Several studies showed an increase in PD along with type 2 diabetes [10,78-81]. However, other studies do not show this correlation [82,83]. Diabetes itself may not be the factor, but instead commonalities between PD and diabetes such as abnormal mitochondrial damage and death [84]. While hormonal therapy for menopause showed an increase risk of PD [85-88], other reproductive related instances, including taking contraceptives, and the age of first menstruation, do not [89].
As mentioned above, Parkinson’s disease has also been related to some skin cancers, specifically, cutaneous malignant melanoma (CMM) [90]. The mechanism relating these two diseases us unknown, but the culprit is likely alpha-synuclein. This protein responsible for characterizing PD is also found in CMM tissue [91]. There are also genetic mutations related to PD onset that have been found CMM reports. Specifically, about 45% of CMM cases contain at least one mutation in PARK [92]. However, PD may help reduce the risk of other cancers. Aside from breast cancers and melanoma, other cancers tend to be low in people diagnosed with PD [93,94].
To follow up the risk factors of PD, there are a few known preventative associations with the disease as well [56]. Urate is a wellknown protective agent [95-97]. It is an antioxidant that preserves dopaminergic neurons for a decrease in their death [98,99]. These neurons are known to degenerate due to oxidative stress, hence why the antioxidant of urate is beneficial. The risk of PD was shown to be significantly lower in people with high levels of urate in their plasma [97]. Studies done with gout patients also confirmed this association of lower PD levels [100,101]. However, there is more evidence to support this theory that it is more than a correlation, but possibly causation [56]. The gene SLC2A9 is a transporter for urate, and in patients with a form of the gene giving rise to higher urate concentration, PD onset was much later [102]. People with varying forms of the SLC2A9 gene with lower urate concentrations in circulation showed higher instances of PD. This correlation was also seen in other genes linked to urate [103]. Due to the abundant amount of information regarding urate as a preventative factor for PD, studies have been done to test its efficacy clinically [104]. A randomized, double-blind study showed that inosine was an effective molecule to raise serum levels of urate in patients with PD. The next step of the study is to show its efficacy in delaying, and potentially reversing, the disease in early onset PD patients [56].
Physical activity has also been shown to be inversely related to PD [105-108]. More frequent moderate or vigorous exercise related to a decrease in PD risk. However, one cannot ignore that increased age tends to decrease the amount of physical exercise a person engages in. Therefore, the correlation of lower physical activity and higher PD risk may be more related to aging. Younger people are more active; however, they are also less likely to develop PD because they are young. Older people have more difficulty with physical activity but also see a higher risk due to their age. However, some argue this, suggesting that the increase in physical shown to be beneficial occurs during early adulthood [105,107].
Certain drug usage has also been associated with decreased PD risk [109]. Ibuprofen has specifically been shown to have an inverse relationship with PD. Patients with higher use of ibuprofen showed a decreased risk of PD. However, other NSAIDs did not follow this pattern [110-112].
The use of tobacco products, including smokeless tobacco, showed a decreased risk of PD [113-115]. Other related instances have been studied such as genetic predispositions to want to smoke that may also lead to PD. However, no correlations with this or personality traits have been found that may explain the connection [116]. Experiments have been done on animal models of PD with nicotine that show healing properties of neurons [117]. However, the other ingredients of tobacco cannot be ignored as having a role in this relationship between tobacco and PD [56].
Coffee consumption, specifically the intake of caffeine, has been shown to lower the risk of PD. This association was seen to a greater degree in men than women. This could be due to an interaction between the caffeine and postmenopausal hormone therapy [118]. Although not every study has supported this relationship between caffeine and low PD risk, these differences are attributed to the setup of the studies not accurately representing the portion of patients that did not consume coffee [119]. Tea consumption has also been attributed with a lower risk of PD compared to people that do not drink tea [120]. However, a study conducted in Singapore showed that black tea had these protective effects but not green tea [121]. These effects are not due to the caffeine content in the tea.
When attempting to diagnose a patient, clinicians look for the typical motor symptoms and non-motor symptoms. These are called supportive criteria, and lead to the diagnosis of PD. Clinicians also wants to see a positive effect of levodopa treatment. However, clinicians also have to rule out any other underlying conditions or diseases that may appear as Parkinsonism. These are called exclusion criteria and include dementia due to Lewy bodies, supranuclear palsy, cerebral tumors, and no effect of high doses of levodopa treatment [13] (Figure 1).
Figure 1: Factors that increase and lower the risk of PD. Taken from “Parkinson’s Disease” [10].
Today’s main treatments consist of controlling the side effects of PD. There are no treatments that stop or reverse the disease [122]. One treatment approach is to replace the dopamine that is lost [123], and another is dopamine agonists, which increase the sensitivity of dopamine receptors [124]. Because of the blood brain barrier, dopamine is incapable of passing from circulation into the brain on its own. Therefore, the drug used to replace lost dopamine is levodopa. L-dopa is a precursor of dopamine that can pass the blood-brain barrier and then be converted into dopamine. This treatment has shown to decrease most motor symptoms. However, not every patient responds as effectively, if at all [125]. There have also been numerous side effects related to L-dopa and other treatments. Side effects of Levodopa include nausea, daytime sleepiness, and fluid retention leading to swelling. L-dopa can also affect the patient’s impulse control leading to gambling, binge eating, and excessive money spending. These side effects are not only found with Levodopa treatment, but also most dopamine agonists. Another frequent side effect seen with dopamine agonists is hallucinations which are a major concern of physicians when prescribing treatments to elderly patients, and they are typically only prescribed to younger patients. While Levodopa shows the most improvement in motor symptoms, it can also induce others, such as dyskinesia, if taken long-term [10].
Lithium has been shown to have more benefits than just a moodstabilizer. Research has shown that lithium plays a role in the nervous system and could be used as a preventative measure for neurological diseases [126]. N-acetyl-aspartate (NAA) is found in the brain and is used as a biomarker for the functionality of neurons. Using quantitative proton magnetic resonance spectroscopy, scientists can measure the amount of NAA in the cortex. Scientists used this method to compare the amounts of NAA after chronic lithium treatment in bipolar disorder patients as well as healthy controls. In both sets of people, NAA increased, showing an increase in viable neurons in that area [127,128]. These results are said to be independent of prophylactic treatment of lithium. Forlenza, et al. [129] conducted a study to see lithium’s effects on the glucose metabolism of neurons in specific areas of the brain. In this study 19 patients to levels of lithium lower than the therapeutic range and hypothesized that the metabolism would increase. Using 18 fluorine-fluoro-2-deoxy-d-glucose positron emission tomography, the metabolism of 12 of the patients showed to be decreased compared to controls; this was not due to lithium toxicity. Therefore, the neuroprotective effects of lithium could not be due to increased metabolism of neurons.
The Lithiumeter shows the plasma concentration levels of lithium for treatment of bipolar disorder. Too low concentration may not be effective or send a patient into relapse. Too high concentration will become toxic. Frequent monitoring is necessary. Figure 2 from “The use of lithium for the treatment of bipolar disorder: Recommendations from clinical practice guidelines” [8].
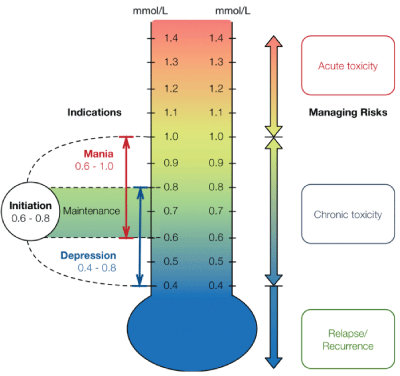
Figure 2: The Lithiumeter shows the plasma concentration levels of lithium for treatment of bipolar disorder. Too low concentration may not be effective or send a patient into relapse. Too high concentration will become toxic. Frequent monitoring is necessary. Figure from “The use of lithium for the treatment of bipolar disorder: Recommendations from clinical practice guidelines” [8].
Lithium has the ability to increase maintenance and function of mitochondria [130], the expression of genes coding for myelination of neurons, and has also been shown to inhibit glycogen synthase kinase-3 [131]. This enzyme (GSK3) is found to have two isoforms, alpha and beta. GSK3 works by recruiting and phosphorylating proteins. GSK3 is well known for over-phosphorylating tau, a protein responsible for tangles found in Alzheimer’s disease [132]. Over 100 substrates have been shown to be phosphorylated by GSK3, and that number is expected to increase as research continues. GSK3 can be inhibited by having its serine-21(in the alpha isoform) or serine-9 (in the beta iso form) phosphorylated. This causes its N-terminal tail to fold into the substrate binding position, blocking other substrates from being able to bind and becoming phosphorylated [133]. Lithium also has a second way of decreasing the activity of GSK3. Lithium binds to a Mg2+ binding site on GSK3, causing lithium and magnesium to compete. GSK3, like many enzymes, cannot work without Mg2+ bound [134]. GSK3β also plays a role in Wnt signaling pathways [135]. In a Wnt pathway, a scaffold is made that requires GSK3β. This scaffold ultimately causes the degradation of β-catenin unless Wnt signals are present [136]. Wnt/β-catenin signaling is necessary early in development of the nervous system, specifically the formation of the neural tube and neural crest [137]. Inhibitors of GSK3β promote neuronal outgrowth, the formation of axons, and an increase in branching of axons [138]. Bcl-2 is a protein highly involved in the regulation of apoptosis as well as the differentiation of neural progenitor cells [139]. Studies have shown that chronic exposure to lithium increase the production of Bcl-2 [140,141].
Lithium was first seen to have beneficial effects towards Parkinson’s disease when neurons were subjected to toxins that resembled PD [142]. They noticed that normally in PD, these toxins cause neurons to undergo apoptosis. When GSK3 inhibitors were introduced, apoptosis decreased. Other scientists have shown that lithium acts to prevent apoptosis through another mechanism as well [142]. Experiments recreating PD neurons in rats showed that lithium inhibited caspase-3, an enzyme causing apoptosis. PD has also been shown to be a related to the amount of oxidative stress that neurons undergo. Scientists have demonstrated that chronic low doses of lithium decrease the negative effects to the cell from oxidation by inducing stress genes [143].
Lithium is also known for its ability to induce apoptosis when necessary via interacting with Bcl-2-associated X protein (Bax) [7]. This protein is responsible for binding to Bcl-2, then p53 induces cell death because of damage. Studies using rats induced with MPTP showed that a diet rich in lithium was able to prevent the increased loss of dopaminergic neurons and only a small amount of dopamine in the striatum decreased [144]. This result was theorized to be due to an increase in Bcl-2 and a decrease in Bax. Similar changes related to Bcl2 have been recorded in patients taking lithium treatment for bipolar disorder, specifically an increase in Bcl-2 in peripheral blood [145].
Lithium is capable of increasing autophagy of cells which helps clear alpha-synuclein [146]. However, studies have shown that this process is decreased in PD patients [147]. When 1,4,5-triphosphate (IP3 ) is created from phosphatidylinositol 4,5-bisphosphate (PIP2), this causes calcium to be released from the endoplasmic reticulum [148]. This causes Ca2+-calpain-G-stimulatory protein alpha (CSα) to be cleaved which inhibits autophagy. Therefore, if IP3 can be decreased, more calcium will be retained, and autophagy can be induced. Lithium has been shown to increase autophagy via this pathway [149]. However, lithium also interacts with many other cellular processes and carries its own side effects when used chronically, causing it to be questioned as a treatment for PD. Studies using rotenone-induced PD in dopaminergic cells showed that lithium was able to reduce the toxic effects of rotenone by increasing autophagy mechanisms [150].
Despite the benefits of lithium therapy, negatives are associated as well. Lithium neurotoxicity has previously been reported. In a review by Bell, et al. [151], patients with previous abnormalities on electroencephalograms (EEG) seemed to show an increased risk of neurotoxicity due to lithium treatment [152]. Lithium treatment also seemed to worsen epileptic episodes in patients with epilepsy [153]. Researchers believe that there is a genetic component to the uptake of lithium in the brain, and some people are at a genetic predisposition to take up lithium more easily [154]. Previously damaged brain matter is also not as effective at removing lithium as healthy tissue [156]. The four cases covered in this review all showed lithium neurotoxicity within therapeutic levels in the serum, and all symptoms of the neurotoxicity were reversed [151]. However, studies have shown that serum levels do not always accurately correspond to levels of lithium in the brain [156]. Levels continue to rise for another 12-14 hours after an initial 12-hour measure of the serum. Instead, more accurate levels of lithium in the brain are portrayed by intra-erythrocyte levels [154].
Although none of the cases discussed showed signs of parkinsonism, people that have damaged brain tissue due to PD may be more prone to neurotoxicity due to lithium because of reduced lithium that is released. Chronic lithium exposure has also been related to cogwheel rigidity [157]. Passive movement around a joint seems to catch and then continue on. Other parkinsonism symptoms such as postural tremor have been noted after chronic exposure to lithium carbonate [158]. A separate incident in 1994 consisted of a man showing signs of lithium neurotoxicity due to lithium treatment for mania [159]. He was subjected to regular serum monitoring of lithium levels, but eight years later showed signs of parkinsonism. However, after passing of acute myocardial infarction, his post mortem analysis did not show any signs of Parkinson’s disease or Alzheimer’s disease, but was suggestive of continued lithium neurotoxicity. This patient could have been genetically predisposed to retain lithium.
Physicians have noticed that children that undergo cranial radiation, usually due to a brain tumor, experience a cognitive decline [160,161]. The effects of radiation in younger patients seem to be worse than adults, which is probably due to the higher turnover rate of neural stem progenitor cells (NSPC) in the hippocampal dentate gyrus in a growing, younger brain [162-164]. Zanni, et al. [165] conducted research to determine the effect of lithium chloride on NPSC in vitro. Other previous pre-clinical data showed that chronic lithium protects against harmful effects of radiotherapy by its anti-apoptotic, anti-inflammatory, and pro-neurogenic capabilities [166,167]. They decided to use 1 mM and 3 mM of lithium chloride on the cells. Their reasoning for determining these concentrations were based on previous work that showed that lithium accumulates in higher levels in cell bodies than what is represented by the therapeutic levels in the blood [168,169]. Their work showed that the cells were able to shorten the G1/S phase transition which led to increased proliferation, the lithium did not affect their ability to differentiate, and the rate of apoptosis was not decreased. Due to these results, lithium treatment given along with cranial radiotherapy is supported in young patients.
A study conducted by Wang, et al. [170] found that lithium chloride assisted in the formation of induced pluripotent stem cells (iPSC) by aiding the reprogramming stage, not by enhancing proliferation. Their results also supported the idea that lithium can be used to aid in the induction of human pluripotent stem cells. Lithium has been shown to have a positive effect to other types of stem cells as well, specifically the differentiation of mesenchymal stem cells (MSC) into neural cells [171]. Green fluorescent protein (GFP)-MSCs were cultured in neural induction medium. Lithium treatment added at 0.1 mM was sufficient to differentiate these cells into neural cells. These GFP-MSCs have also been implanted into rats and then had the lithium treatment add in vivo. The results showed that the cells were viable and still differentiated into neural cells.
Neural stem cells have also been shown to be positively affected by lithium treatment, specifically in its use for Alzheimer’s disease treatment and aid of neurogenesis and improved cognitive function [172]. A main source of neurogenesis is found in the neural stem progenitor cells, and lithium is able to stimulate their proliferation [9,173]. Previous studies have been done to observe the effects of lithium on adult neural progenitor cells and their increasing ability to proliferate, differentiate, mature, and function, due to lithium [174-177]. Lithium has also been shown to reduce the inflammatory responses caused by microglial cells in the brain of mice [178]. Microglia induced inflammation is believed to be a source for the beginning of degeneration of neurons in early PD, making lithium’s ability to reduce this a possible key to PD delay
Research shows that individuals diagnosed with BD are at an increased risk of dementia. However, when undergoing lithium therapy for BD, the risk of dementia drops down to the level seen in the general population. The risk of dementia increases with each episode a BD patient experience. This specific study showed that this risk also decreased when lithium treatment continued. Gray matter also increases in as few as 4 weeks of lithium treatment [179].
Lithium seems to particularly have many benefits to neurological diseases. This could be due to the fact that it is an anti-inflammatory agent, which is a key characteristic of these diseases. Neurological diseases have also been shown to cause a decrease in BDNF. This protein is part of a signaling process that helps to inhibit GSK3. If BDNF is in low levels, this could lead to the over activation of GSK3, giving rise to these diseases that are related to GSK3 [180].
The immune system is also positively affected by lithium [181]. Monocytes are a type of white blood cell found in circulation that eventually takes residence in tissues as macrophages. A study was conducted that determined that human BD patients treated with lithium carbonate showed an increase in the amount of TNF-alpha that was secreted by their monocytes [182]. A separate study was conducted to see the effect of lithium on hematopoietic stem cells (HSC). Patients underwent lithium treatment for 3-4 weeks and showed a significant increase in the number of neutrophils, as well as CD34+ cells [183] which is a protein found on the surface of HSCs [184]. Lithium has since been used as a pre-treatment for bone marrow transplants to increase the desired HSC [185]. Lithium has been shown to increase the production of antibodies, specifically IgM and IgG. However, it has also been shown to decrease production of c-AMP. Researchers are uncertain if these situations are related [186]. Experiments have been done showing that cell cultures treated with lithium chloride suppresses herpes simplex virus-1 mRNAs, and the viruses are unable to replicate [187].
Similar to any drug, lithium has to be taken carefully, and there are common side effects seen in a small cohort of patients. Some side effects seen at maximum safe levels are excessive urination volume (polyuria), tremors, and cognitive impairment. A level of 1.5 mmol/L begins toxicity, and above 2.0 mmol/L needs medical attention. Low dose formulations have made toxicity less of an issue when taken correctly. Taking lithium chronically can also cause problems with the endocrine system and kidneys. An attempt to circumvent these issues can be made by regular blood monitoring. Doctors should have a measurement of the background levels of lithium found naturally in the patient’s blood before treatment begins. Monitoring should then continue in a regular pattern to see how the body handles dosages so that lithium serum levels fall with the therapeutic range [8].
Women that plan to become pregnant, or are pregnant, and undergoing lithium treatment should talk to a physician weighing the pros and cons of continuing treatment during pregnancy. There is a potential for teratogenicity during the first trimester, and this should be weighed against stopping treatment altogether for the mother which could cause relapse. A possibility is to decrease the dosage so that lithium serum levels are lower. After a woman gives birth, breastfeeding should be avoided [8].
Cell therapy for PD originated with trials using fetal cells from the substantia nigra of mice. Grafts were taken from fetal mice that included dopaminergic neurons [188]. When implanted into mice simulating PD, motor symptoms improved. The grafts were sustained and created new synapses with the cells already existing. Aside from the potential immunological and ethical issues, the studies showed positive results in motor function and that the procedure was safe. Two clinical trials were done in 1988 which showed unwanted dyskinesias in elderly patients, and only some efficacy in younger patients [189,190]. Trials using this source of cells for grafts began to slow. Eventually, further research using the same idea, but with the cells in suspension, were shown to be another possible avenue for PD therapy [191]. A few patients showed improvement in their motor symptoms. Additionally, no adverse effects were seen, despite the low use of immunosuppressants. Others even showed improvement for up to ten years. Fetal cell transplantation has shown to have a positive outcome in the few patients that have been tested [192]. A significant study is currently being conducted in Europe, TRANSEURO, and the results are highly awaited. Still, other areas of research are being done to circumvent the ethical issues of using aborted fetus cells.
A clinical trial has been done using an autologous transplant of cells from the carotid body in the carotid artery [193]. In development, these cells differentiate from the neural crest and produce glial cell linederived neurotrophic factor (GDNF) which acts as a neuroprotective agent to the transplant. While the clinical trial showed positive results in most of the patients and no negative effects, there is not much research that has been done in this area.
In 2000, Kawasaki cultured embryonic stem cells (ESC), along with their stromal cells, and was able to differentiate the ESCs into cells similar to dopaminergic neurons [194]. ESCs have been shown to be a promising candidate for PD therapy, but research has moved on to other sources of cells that circumvent the ethical issue [192].
Breakthrough research happened in 2006 when Yamanaka made the first induced pluripotent stem cells (iPSC) using skin fibroblasts [195]. He added a specific combination of OCT4, SOX2, KLF-4, and c-Myc to the fibroblasts. This enabled the fibroblast cells to differentiate into cell types from each of the three germ layers. This is a characteristic of embryonic stem cells. There were noticeable differences, however, between Yamanaka’s cells and ESCs, specifically in their mechanisms for proliferating. The iPSCs were implanted into a blastocyst, and they did not form a viable embryo, adding another difference from ESCs. Although iPSCs cannot be considered identical to ESCs, they contain the same differential ability as ESCs and are pluripotent [196]. These types of stem cells are very appealing to scientists because of their differentiating capabilities while remaining ethical. Many different types of cells can be used as a starter to create iPSCs including cells from the stomach, liver, neurons, and even blood cells. Because of how minimally invasive collecting a blood sample is, these cells are the most alluring as the source for creating iPSCs. Researchers have shown that as little as 10 μL of blood from a fingertip contains a sufficient amount of blood cells to use to create iPSCs [197].
This new line of stem cells can also be used to model diseases to gain knowledge and test drugs for specific patients. Most studies using iPSCs from PD patients being used to study the genetic component of the disease focus on the G2019S mutation in LRRK2, which is the most common mutation in familial cases of PD [198]. This study showed that these cells are especially susceptive to oxidative stress compared to controls. Gene editing has been tested in these types of cells and this same mutation, G2019S [199]. Phenotypes expressed by PD, such as vulnerability to oxidative and mitochondrial stress, were improved. iPSCs have also been used to create three-dimensional (3D) organoid systems. These systems are better at mimicking situations in vivo. The same mutation in LRRK2 that has been studied in 2D cell cultures has also been studied in 3D organoids [200]. Gene expression differences were noticed that corresponded to the synaptic transmission in these organoids as compared to controls.
A main concern of using iPSCs as a potential graft source is causing tumors in the patient. Myc and KFL-4 are both oncogenes that when mutated, cause changes in a cell’s metabolism and proliferation which leads to cancer [201]. The other transcription factors and their genes are also associated with cancer. Often when iPSCs are being created, a virus is inserted to the cell, which can disrupt the host genome and affect these oncogenes [202]. Induced pluripotent stem cells can be invaluable to determining the mechanism through which PD develops. iPSCs can be generated from a patient that has PD, and the cells can be manipulated and monitored to see the effects. Research was unsettled on the presence of mitophagy in PD rat models. Healthy cells have to undergo mitophagy to get rid of the ones that are damaged [18]. During mitophagy, an autophagosome engulfs the mitochondrion and then fuses with a lysosome so that it can be degraded by enzymes. Researchers found that a kinase specific for the mitochondrion, PTEN-induced kinase 1 (PINK1), builds up on the outside of damaged mitochondria [203] in neurons. This accumulation recruits Parkin which tags the organelle with ubiquitin for its degradation. Still, other scientists showed that Parkin was not recruited in mice models of PD [204]. Using iPSCs, the PINK1 gene was mutated and showed less recruiting of Parkin and mitochondrial breakdown. However, another factor was shown to cause mitophagy besides PINK1/Parkin. Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) also induces mitophagy. The CCCP-induced mitophagy is thought to be independent of PD, but it may lead to mitochondria that are not autophaged. Rather, they are left damaged, still able to provide a small amount of energy for the cell. This is due to CCCP causing the mitochondrial membrane to become more permeable, allowing protons to flow in, damage the membrane’s potential, and possibly causing apoptosis. Using iPSCs, this mechanism can be further understood [196]. These types of stem cells may also be beneficial in finding PD early. Characteristics of cells in PD are mitochondrial damage, oxidative stress, and other cellular functions, may act as markers for PD before it is noticed through the main symptoms [196].
Using iPSCs as a source for cell grafts has been a major area of research [205,206]. Previous studies using the fetal cells showed the importance of purified grafts, as to avoid unwanted dyskinesias due to serotonin-producing cells in the graft [188,205]. Cell sorting can be done with certain biomarkers, such as NCAM+/CD29low which improved the quality of the graft and its efficacy. This screening for certain markers can also weed out any tumor cells potentially causing cancer. The suggested number is 100,000 dopaminergic cells in the graft to be successful, and studies have shown expansion of the grafted neurons if the grafts contain enough neural progenitor cells. The environment in which somatic cells are induced is also extremely important. Research shows that rosette-cultured cells have higher proliferation than floor plate. Using the patient’s own cells for the transplant seems the best in relation to histocompatibility. However, if the cells are still affected by the disease, due to genetics, this would not solve the problem. In this case, another person that is HLA-matched would need to be considered. Gene editing is available; however, it would not be ideal as there are many factors leading to PD than a single genetic mutation [208].
The studies with the fetal mesencephalic tissue showed improvement in a patient’s motor symptoms; however, they failed to treat non-motor symptoms that eventually appeared. This tells that the graft is unable to stop PD from progressing further. Once the disease has progressed into parts of the brain not relating to the dopamine system, a graft of dopaminergic cells is not effective. Patients that respond positively to levodopa drug treatment would be good candidates for a cell graft. Because PD patients with genetic PD do not show many motor symptoms, these people may also be more responsive to dopaminergic cell grafts [196]. The microenvironment of the graft is also very important. If the surrounding cells contain Lewy bodies, these proteins can migrate into adjacent cells, potentially staring Lewy bodies in the healthy grafted cells [209].
Mesenchymal stem cells (MSC) are easily obtained in significant quantities and have been shown to be re-programmed. Specifically, Dezawa induced MSCs into neuronal cells by gene transfection and specific growth factors and chemical signals [210]. The amount of glial cell line-derived neurotrophic factor (GDNF) was related to the ability of the cells to differentiate into dopaminergic neurons: more GDNF, more dopamine producing neurons. However, even with these promising outcomes, no clinical trials have shown beneficial results with PD using MSCs [192]. Venkataramana used MSCs from the bone marrow of an autologous donor and implanted them into the sublateral ventricular zone. The results seemed slightly promising as motor function improved in a number of the patients. However, the study lacked controls and the sample size was small, so the trial did not show efficacy [211].
As Parkinson’s disease is not fully understood, treating the disease can be difficult, while preventing, and reversing it is still impossible. The current main treatment for motor symptoms is levodopa drugs to replace lost dopamine, or receptor agonists to increase their sensitivity. Mitochondrial and autophagy dysfunction are underlying mechanisms found throughout PD studies. Lithium could be used as a preventative measure to block GSK3 from hyperactivation, and lithium has also been shown to increase autophagy and assist mitochondrial processes. Lithium treatment also protected cells against rotenone-induced PD. Somatic cells of PD patients could be induced to iPSCs to study the progression of the disease in 2D or 3D model organoids. Cell therapy has been used in grafting of dopaminergic neurons into affected areas of the brain. The history began with fetal mesencephalic tissues and has led currently to iPSCs. Lithium has also shown to be promising in the area of iPSCs. While MSC therapy has not grown significantly as it relates to PD, the results of lithium promoting MSC differentiation into neural cells are promising for MSC grafts. Special consideration must take place concerning the advantages and disadvantages of the current therapy using levodopa with stem cell grafts. Because the grafts would be most efficient in patients with less non-motor symptoms, levodopa is typically extremely effective. Do the benefits from the graft outweigh the risks of the surgery when levodopa might lead to those same benefits? Stem cells from PD patients are, however, invaluable in determining the mechanisms through which PD develops, showing a promising future in answering the unknowns of PD.
Download Provisional PDF Here
Article Type: REVIEW ARTICLE
Citation: Burckhalter L, Gallicchio VS (2019) Parkinson’s disease, Lithium and Stem Cells. J Clin Lab Med 4(1): dx.doi.org/10.16966/2572- 9578.126
Copyright: © 2019 Burckhalter L, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access