Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a hyper inflammatory condition caused by a highly stimulated and defective inflammatory response. The underlying diseases are various, including infections, autoimmune diseases, and cancer, especially lymphomas and leukemias. HLH is a fatal disorder and the early diagnosis is required. It is also important to identify the underlying disease and to start an appropriate treatment as soon as possible.
Here, we present a case of aggressive natural killer-cell leukemia (ANKL) with HLH that was diagnosed promptly by flow cytometry (FCM) analysis.
A male patient in his sixties was admitted to a hospital due to persistent fever, general malaise, hepato-splenomegaly, and thrombocytopenia. A bone marrow aspiration was performed and acute lymphocytic leukemia was suspected. He was then referred to our hospital for advanced care. His complete blood cell count showed normal range of WBC (3.7 ×109 /L) with 3% of atypical lymphocytes and thrombocytopenia (18×109 /L). His second bone marrow test revealed 64.9% of macrophages with hemophagocytosis of granular blastoid cells. Although the morphological diagnosis was difficult due to massive hemophagocytosis in bone marrow, the blastoid cells were characterized by FCM analysis. The abnormal population was positive for CD2, CD7, CD16 and CD56. His laboratory data showed hypofibrinogenemia and hyperferritinemia as well. He fulfilled 5 of 8 HLH criteria. Therefore, ANKL with HLH has been diagnosed.
Keywords
Hemophagocytic Lymphohistiocytosis; Natural killer (NK) cell; Aggressive NK cell Leukemia; Flow cytometry
Introduction
Hemophagocytic lymphohistiocytosis (HLH), also hemophagocytic syndrome (HPS), can be triggered by infections, autoimmune or malignant diseases [1]. The diagnostic criteria of HLH were proposed by the Histiocyte Society HLH-2004 protocol [2,3]. The diagnosis of HLH can be established by: A. a molecular diagnosis consistent with HLH or B. five of 8 criteria listed below are fulfilled; 1) Fever; 2) Splenomegaly; 3) Bicytopenias (hemoglobin <9g/dL, neutrophils<1.0 × 109 /L, platelets <100 × 109 /L); 4)Hypertriglyceridemia (>265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL); 5) Hemophagocytosis in bone marrow or spleen or lymph nodes or liver; 6) Low or absent NK-cell activity; 7) Ferritin >500 ng/mL; and 8) Elevated Soluble CD25>2400 U/ mL. HLH is induced by a highly stimulated and defective inflammatory response involving 3 main pathways; i.e 1) hyperactivation of CD8+ T lymphocytes and macrophages, 2) proliferation and infiltration of these cells into organs and tissues, and 3) uncontrolled hypercytokinemia [4-6].
Here we present a case of secondary HLH associated with a malignant disease. The patient’s bone marrow test showed massive hemophagocytosis of blastoid cells by macrophages. We were not able to characterize the blastoid cells morphologically. However, those blastoid cells were identified as natural killer (NK) cells through antigen detection on their cell-surface by flow cytometry (FCM), and the patient was diagnosed as aggressive NK-cell leukemia (ANKL) accompanied by HLH.
Case
A Japanese male patient in his sixties was admitted to a neighborhood hospital due to fever and general malaise. He also presented with increased level of transaminase, lactate dehydrogenase (LDH), and total bilirubin. He was diagnosed with possible acute cholecystitis. Although he received antibiotics therapy, the symptoms did not improve. Because he also developed thrombocytopenia, a bone marrow aspiration was performed that revealed 91.8% myeloperoxidase-negative blastoid cells. The patient was then referred to our hospital for advanced care for the possible diagnosis of acute lymphocytic leukemia.
His laboratory data are shown in (Table 1). Complete blood count revealed a marked decrease of the platelet count (18 × 109 /L). Hemogram showed 3% of atypical lymphocytes with erythroblastosis. The calculated DIC score by the International Society of Thrombosis was 6, which made DIC probable. Blood biochemistry results showed liver injury pattern including increased transaminase (AST 709 U/L, ALT 283 U/L), LDH (4530 U/L), direct-bilirubin (4.4 mg/dL), and alkaline Phosphatase (990 U/L). The serum total cholesterol (98 mg/dL) and HDL cholesterol (5 mg/dL) levels were low, while the serum ferritin (13300 ng/mL), soluble interleukin-2 receptor (1112 U/mL) and thymidine kinase (1264.7 U/L) levels were increased. His abdominal computed tomography scan showed hepato-splenomegaly and para-aortic lymphadenopathy.
+
| Test |
Results |
Unit |
Normal range |
| red blood cell |
4.58 |
x1012/L |
4.35-5.55 |
| hemoglobin |
14.1 |
g/dL |
13.7-16.8 |
| hematocrit |
42.6 |
% |
40.7-50.1 |
| platelet |
18 |
x109/L |
158-348 |
| white blood cell |
3.7 |
x109/L |
3.3-8.6 |
| Hemogram |
|
|
|
| myelocyte |
5 |
% |
0 |
| neutrophil |
58 |
% |
40-69 |
| lymphocyte |
33 |
% |
21-35 |
| monocyte |
1 |
% |
4-8 |
| atypical lymphocyte |
3 |
% |
0 |
| erythroblast |
42/100WBC |
|
0 |
| PT* |
20.6 |
seconds |
No settings |
| PT% |
36.0 |
% |
80-100 |
| PT-INR* |
1.83 |
|
No settings |
| APTT*` |
58.1 |
seconds |
24.5-33.8 |
| fibrinogen |
103 |
mg/dL |
200-400 |
| antithrombin |
46 |
% |
80-130 |
| SFMC* |
15.6 |
µg/mL |
6.1> |
| TAT* |
46.2 |
ng/mL |
3.0> |
| FDP* |
7.5 |
µg/mL |
5.0> |
| D-dimer |
5.6 |
µg/mL |
1.0> |
| PIC |
1.2 |
µg/mL |
0.8> |
| total protein |
4.6 |
g/dL |
6.6-8.1 |
| albumin |
2.9 |
g/dL |
4.1-5.1 |
| T-Bil* |
6.5 |
mg/dL |
0.40-1.50 |
| D-Bil* |
4.4 |
mg/dL |
0.03-0.40 |
| AST* |
709 |
U/L |
13-30 |
| ALT* |
283 |
U/L |
10-42 |
| LD* |
4530 |
U/L |
124-222 |
| ALP* |
990 |
U/L |
106-322 |
| γ-GT* |
321 |
U/L |
13-64 |
| T-CHO* |
98 |
mg/dL |
142-220 |
| HDL-CHO* |
5 |
mg/dL |
40-90 |
| triglyceride |
185 |
mg/dL |
40-150 |
| uricacid |
10.5 |
mg/dL |
3.7-7.0 |
| urea nitrogen |
15.2 |
mg/dL |
8.0-20.0 |
| creatinine |
0.82 |
mg/dL |
065-1.07 |
| blood sugar |
54 |
mg/dL |
73-109 |
| sodium |
130 |
mEq/L |
138-145 |
| potassium |
5.7 |
mEq/L |
3.6-4.8 |
| chloride |
92 |
mEq/L |
101-108 |
| calcium |
8.2 |
mg/dL |
8.8-10.1 |
| ferritin |
13300 |
ng/mL |
26.0-388.0 |
| sIL-2R* |
1112 |
U/mL |
145-519 |
| thymidine kinase |
1264.7 |
U/L |
7.5> |
| ammonia |
158 |
µg/dL |
12-66 |
Table 1: Laboratory data on admission
*Abbreviations: PT (prothrombin time) INR (international normalized ratio), APTT (activated partial thromboplastin time), SFMC (soluble fibrinmonomer complex), TAT(thrombin-antithrombin complex), FDP (fibrin/ fibrinogen degradation products), PIC (plasmin-α2 plasmin inhibitor complex), T-Bil(total bilirubin), D-Bil (directbilirubin), AST (aspartate transaminase), ALT (alanine transaminase), LD (lactate dehydrogenase), ALP (alkaline phosphatase), γ-GT(γ-glutamyltransferase), T-CHO(totalcholesterol),HDL-CHO(highdensity lipoprotein-cholesterol), sIL-2R (soluble interleukin-2receptor)
We repeated the bone marrow test. The blastoid cells that were observed in the previous bone marrow test were no longer seen in a countable form; macrophages accounted for 64.9% of the total nuclear cells, exhibiting hemophagocytosis (Figure 1). Aggregated cells were sporadically observed at a magnification of X100. At a magnification of X1000, there were macrophages phagocytosing various blood cells including some large blastoid cells containing coarse azurophilic granules in their cytoplasm in places. We were not able to characterize those blastoid cells morphologically.
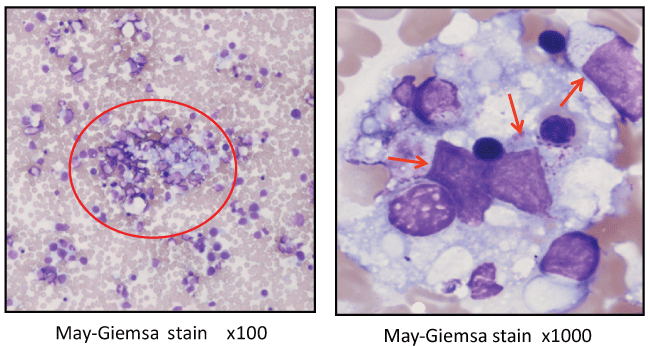
Figure 1: Bone marrow smear showing macrophages exhibiting hemophagocytosis. Aggregated cells were sporadically observed at a magnification of X100 (left). At a magnification of X1000 (right), there were macrophages phagocytosing various blood cells including some large blastoid cells containing coarse azurophilic granules in their cytoplasm in places.
The results of cell-surface antigen analysis by FCM are shown in (Figure 2). The mature cell population showing strong surface expression of CD45 was detected by the CD45 Blast Gating method. This cell population was positive for CD2 and CD7 (T cell lineage), CD16 and CD56 (NK cell lineage), and CD64 (macrophage and monocyte lineages), but negative for surface CD3, CD4 and CD8. A noteworthy finding in the emergence of the cell populations is that the fluorescence intensity distribution generally separates the cells into lymphocytes showing weak side scatter (SS) and monocytes showing relatively strong SS; however, these two clusters were huddled into one population in this case (Figures 2A and 2B).
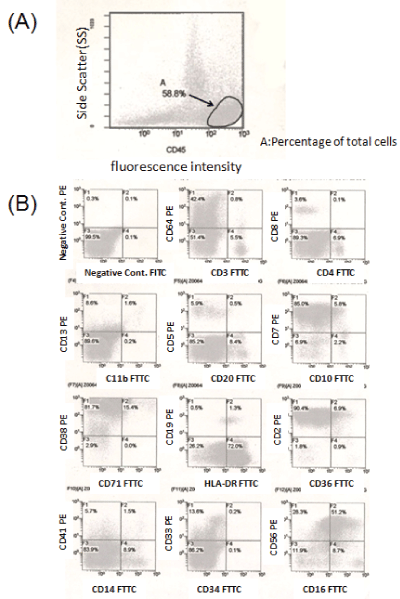
Figure 2: The cell-surface antigen analysis by FCM (A): The mature cell population showing strong surface expression of CD45 was detected by the CD45 Blast Gating method. (B): This cell population was positive for CD2 and CD7 (T cell lineage), CD16 and CD56 (NK cell lineage), and CD64 (macrophage and monocyte lineages).
Chromosomal analysis revealed evidence of Chromosome 8 trisomy in 2/20 cells (Figure 3).The patient fulfilled 5 out of the 8 HLH criteria: fever, splenomegaly, hypofibrinogenemia, hyperferritinemia, and hemophagocytosis in bone marrow. Collectively, we diagnosed the patient with ANKL accompanied by HLH on his admission day.
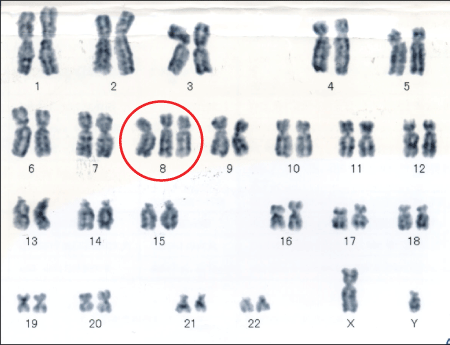
Figure 3: Chromosomal analysis showing trisomy 8.
Despite the prompt diagnosis and treatment, he died 16 days after admission to our hospital due to bacteremia and gastrointestinal bleeding.
Discussion
ANKL is a specific type of leukemia, in which mature NK cells proliferate throughout the body [7-9]. The disease may manifest with decreased blood cell counts and hepato-splenomegaly. DIC and HLH are known complications of ANKL. There are several cell forms peculiar to this disease, such as ones that cannot be distinguished from normal granular lymphocytes or relatively large cells. They may have irregular nuclei, and many of their nuclear reticulum shows aggregated chromatinpattern. Fine or coarse azurophilic granules are found in the cytoplasm [7-9]. In regard to the cell surface immunophenotypes, the cells are positive for CD2, CD16 (sometimes negative) and CD56 and negative for surface CD3 [7].
In our case, the bone marrow smear showed hemophagocytosis; however, it was difficult to define the underlying disease. Although there were cells with characteristics similar to those of NK cell in the bone marrow smear, they were found only occasionally and most were found in a phagocytosed form. Since cell populations clearly showing the surface antigen profile of cells involved in ANKL were observed by FCM, the blastoid cells with granules could be identified as NK cells. Considering reports that ANKL can be complicated by HLH [7,8,10] and considering his laboratory findings the patient was diagnosed having ANKL. Although approximately 90% of blastoid cells were found in the first bone marrow test, probably most of them had been phagocytosed by the time the patient was transferred to our hospital. The phagocytosis of the malignant NK cells by macrophages could be the reason for the lack of separation of the lymphocyte and monocyte lineages in the FCM analysis.
Since emerging locations of tumor cells vary depending on the form of the disease or case, in general, gating after identifying the cell forms is very important for diagnosis. In this case, on the contrary, since the FCM analysis indicated a possibility of existence of tumor cells, the bone marrow sample was carefully examined again, which revealed the NK cells admixed with the macrophages.
No chromosomal abnormalities specific to ANKL are believed to be known; however, some have pointed out del (6) (q21q25), deletion of 11q in this disease [11,12], and complex karyotypes [13], including chromosome 1 and 8 abnormalities [10,14].The chromosomal 8 trisomy has been found in this case.
ANKL is considered as one of Epstein-Barr virus (EBV) associated malignancies. EBV-infected NK cells constitutively secrete high levels of interferon-gamma (IFN-G) which helps maintain cell survival, and preventing apoptosis via autocrine fashion [15]. The high level of IFN-G might be the trigger for HLH through activation of macrophages and histiocytes. This could be the reason that ANKL is often accompanied by HLH.
Recently more easily and rapidly diagnosis of HLH has been investigated that includes 18 variables; bone marrow/lymph node/ spleen hemophagocytosis per pathology evaluation, fever, splenomegaly, hepatomegaly, thrombocytopenia (<100 × 109 /L), neutropenia (<1.0×109 /L), monocytosis (>1.0×109 /L), renal failure (>50% increase in creatinine over baseline), elevation of hepatic enzymes (>2.5 times the upper limit of normal), hypofibrinogenemia (fibrinogen <150 mg/dL), hyperferritinemia (ferritin >500 ng/mL), coagulopathy (prothrombin time >1.5 times the upper limit of normal, and/or partial thromboplastin time >1.5 times the upper limit of normal, and/or D-dimer >10.0 ug/ mL), hypoalbuminemia (<3.5 g/dL), elevated LDH (>2.5 times the upper limit of normal), hypertriglyceridemia (>256 mg/dL), elevated β2- microglobulin (>2 mg/L), and elevated soluble IL-2 receptor (>2400 U/ mL). Most of these variables are readily and quickly available in regular clinics. When a patient shows more than 5 of 18 variables, the can be considered as having HLH [4-6]. In our case, the patient met out of 18 criteria. Thus the patient fulfilled both HLH-2004 and the new extended criteria. Since HLH is a life-threatening disorder, it has to be diagnosed as quickly as possible. The new extended criteria may be useful for a rapid diagnosis. An investigation of the underlying disease also needs to be performed quickly and FCM analysis could even characterize malignant cells that are being phagocytosed.
Conclusion
We presented a patient who was diagnosed with ANKL accompanied by hemophagocytosis through cell identification by FCM analysis. HLH can occur in various diseases including malignant lymphomas and leukemias. In such cases, obtaining the necessary information for diagnosis is thought to be possible by identifying cell forms as well as examining the results of FCM and analyzing the patient characteristics.
Article Information
Article Type: Case Report
Citation: K Nogi, H Tashiro, K Kawasugi , M Matsuzawa, T Osawa,et al. (2017) Aggressive NK Cell Leukemia with Hemophagocytic Lymphohistiocytosis: A Case Report. J Clin Lab Med 2(2): doi http://dx.doi.org/10.16966/2572-9578.116
Copyright: © 2017 Nogi K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 21 Aug 2017
Accepted date: 19 Sep 2017
Published date: 25 Sep 2017