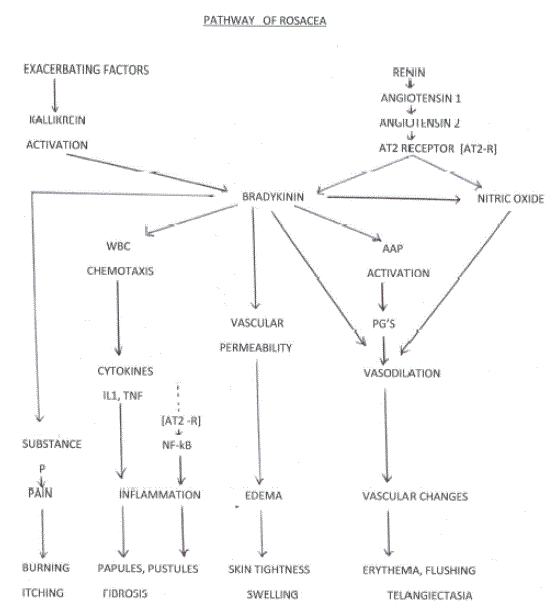
Figure 1: Pathway of Rosacea
IL=interleukin NO=nitric oxide TNF= tumor necrosis factor PG=prostaglandin AAP=arachidonic acid path WBC=white blood cell

John Bolla*
American Board Family Medicine, American Academy Family Physicians, USA*Corresponding author: John Bolla, 3 Wilderness Run, Flagler Beach, Florida, USA, Fax: 32136; Tel: 386-931-5920; E-mail: johnbolla@cfl.rr.com
Rosacea is a disorder the affects millions of people and remains unexplained in terms of its pathophysiology. Many hypothesis have been put forward bun none has led to the breakthrough that so many researchers want. I believe that this is due in part because of the nature of basic research that limits forward movement to small steps, each adding another piece to the puzzle, but not enough to allow for completion of the big picture. I have been fortunate enough to have treated a patient who developed Rosacea as a consequence of another medical problem, thus allowing me to start with the big picture and work back through the pathophysiology and determine how this could have happened. The disorder in question is renovascular hypertension which occurred as a result of an acute renal artery dissection. The renin angiotensin system is activated in this situation and the cascade of events that follows can both initiate and sustain a state of vasodilation in the vasculature of the face that results in the clinical expression of Rosacea. Hypertension and Rosacea are recognized as comorbid conditions that are linked in s severity dependent manner and the hypothesis presented in this paper explains why this is so. It also presents the Angiotensin AT2 receptor as a possible target for therapeutic intervention.
Effective therapy for Rosacea has been described as one of the great unmet needs in dermatology [1]. This is a disease which has eluded talented researchers for many years and continues to present questions which remain unanswered. In a recent review in 2018, the authors concluded that what we need to know most is what the key mediators and receptors involved in Rosacea are [2]. In this paper, I intend to shed light on that question.
I will first review the classification of Rosacea based on the 2004 system [3,4]. I will offer a proposal of what I believe to be the pathophysiology of Rosacea and the clinical case which serves as a model for it. I will detail the research that supports my proposal and tie it together into a unified explanation of this disease. Finally, I will suggest the therapeutic approach that naturally derives from this rationale and thereby bring to the table the possibility of developing a novel and effective therapy for this disease.
Rosacea is a chronic inflammatory disease of the skin characterized by four subtypes:
Subtype 1: Erythematotelangiectatic
Persistent erythema, flushing, telangiectasia
Subtype 2: Papulopustular
Papules, pustules, persistent erythema
Subtype 3: Phymatous
Skin thickening, Rhinophyma
Subtype 4: Ocular
Red irritated eyes
Patients complain of redness, burning, itching, pain, swelling and skin tightness.
Exacerbations are caused by:
Heat and humidity
Sweating
Exercise
Sunlight
Stress (adrenergic stimulation)
Alcohol
Rosacea is an external manifestation of an internal disorder characterized by elevated levels, either local or systemic or both, of Angiotensin II. This causes stimulation of the AT2 receptor, production of bradykinin, nitric oxide, and a cascade of inflammatory mediators in the vasculature of the face which result in a state of chronic vasodilation and inflammation.
The exacerbating factors which cause a worsening of Rosacea do so by triggering activity of the kallikrein enzyme in the skin which then activates bradykinin.
There are thus two arms- an Angiotensin arm and a skin kallikrein arm- that cause vasodilation and inflammation by acting through one common pathway (Figure 1).
Figure 1: Pathway of Rosacea
IL=interleukin NO=nitric oxide TNF= tumor necrosis factor PG=prostaglandin AAP=arachidonic acid path WBC=white blood cell
The patient in question was a 43-year-old male who suffered a spontaneous bilateral renal artery dissection. This is a rare disorder with about 200 cases reported worldwide since 1944 [5]. The dissection resulted in patchy diffuse ischemic renal disease, worse on the right kidney than the left, and stenosis of the right renal artery. He developed acute and severe hypertension and required hospitalization for 12 days at a tertiary facility in the United States. He did well and was discharged home on a beta blocker in a normotensive state. He did not tolerate ACE inhibitors.
The patient had no history of hypertension or any other medical problems and was not on any medications prior to the onset of the event. He did not smoke or take drugs. He did drink alcohol socially. He was athletic and had just finished running when the dissection started. His BMI was 24.
He had no dermatologic issues nor history of any. There was no family history of dermatologic disease. There was a history of hypertension in one parent.
Six weeks later, he began to develop facial erythema, and by 8 weeks it was so noticeable that people were asking him if he had spent the weekend outside without any sunscreen. He could count his pulse by feeling the pulsations in his nose which he said was getting noticeably larger. He found the disorder painful, with a feeling that the skin was being stretched and had been burnt. He could see small blood vessels appearing on his face.
The patient sought help. He was treated with oral tetracycline at 500 mg bid and topical metronidazole bid. This improved nothing over an 8-week period, and he did not tolerate the metronidazole due to burning and aggravation of the Rosacea. He began to develop papules and pustules which did respond modestly to topical sodium sulfacetamide sulfur lotion applied bid. Topical Retin-A was not helpful. Because of the severity, rapidity of development, and poor response to medication, he was referred to a dermatologist. The doctor suggested the combination of tetracycline and sulfa was a good one and it should be continued. He advocated trigger avoidance and basic care of the skin. The patient went for a second opinion with another specialist and was given the same advice along with options of isotretinoin and laser therapy. The patient declined both at that time because of concerns for potentially serious side effects. Neither dermatologist had any ideas regarding the possible connection between the renovascular hypertension and the Rosacea. There were no other drugs available at the time (1998). Azelaic acid and brimonidine were not yet marketed.
The patient’s hypertension moderated with time and he remained on a beta blocker. The Rosacea moderated gradually and was better controlled with doxycycline, topical sulfa, and laser therapy
Renal artery dissection produces secondary hypertension due to extremely high levels of Angiotensin which are produced in this situation. When the renal artery dissects, the layers of the artery separate, platelets adhere, and clots form in the vessel which leads to varying degrees of obstruction. Low pressure in the renal artery is a stimulus for renin production. Renin generates Angiotensin 1 which is subsequently converted to Angiotensin 2. Angiotensin 2 then causes vasoconstriction by acting through the AT1 receptor. This causes secondary hypertension and is what this patient developed.
The existence of Angiotensin receptors other than the AT1 emerged in the late 1990’s. There are a number of these but the most studied is the AT2. It has now been shown that this receptor produces vasodilation by generating bradykinin, which in turn generates nitric oxide [6-9]. Nitric oxide can also be generated directly via the AT2receptor [10].
Bradykinin causes activation of the arachidonic acid pathway [11-16]. The arachidonic pathway generates prostaglandins which themselves cause vasodilation, nitric oxide production, and generation of oxygen free radicals.
Bradykinin also causes chemotaxis of white blood cells and release of cytokines from them, causes an increase in vascular permeability, and causes release of substance P from nerve endings, resulting in pain [17,13].
AT2 receptor stimulation has also been shown to activate NF-kB, a proinflammatory nuclear factor in vascular smooth muscle. This was demonstrated in both invitro and invivo studies and the effect were mitigated by AT2 receptor blockade [18].
Hence, it can be seen that stimulation of the AT2 receptor has the capability to cause all the vascular and inflammatory changes that make up the signs and symptoms of Rosacea (Figure 1).
AT2 receptors have been identified in most tissues in the body and are present in vascular endothelium [6,7,19]. Their numbers are greatest in the fetus and diminish with age. The AT2 receptor can be upregulated in two situations- the first is injury and the second is in the presence of high levels of Angiotensin 2.
Upregulation due to injury has been demonstrated in rat skin [20], rat carotid arteries [19], and in human skin [21]. In human skin the increase was measurable at 24 hours and sustained for three months. What is important to note is that the response is rapid and sustained over months, not hours or days. In addition, the mechanism of injury was not the same in all the studies. Scalpel was used on skin and balloon angioplasty was used to traumatize rat aorta. The fact that different mechanisms can cause upregulation is an important concept that I will come back to later.
Upregulation of AT2 receptors occurs in the presence of elevated levels of Angiotensin 2. This has been demonstrated with Angiotensin 2 infusion [19,22] and with AT1 receptor blockade [23]. Blockade of the AT1 receptor interrupts what is referred to as the “short feedback loop” that controls renin production in the kidney. Normally, AT1 stimulation turns off renin production, but with the receptor blocked, renin continues to cause Angiotensin production and Angiotensin 2 levels rise, producing the same effect as an infusion. Both number and density of AT2 receptors are increased in this situation. The AT1 receptor does not go up in this situation.
In the case presented, the patient would have had extremely high levels of Angiotensin 2 because of the renal artery dissection. This is evidenced by his severe hypertension, a consequence of AT1 receptor activation. One can conclude, therefore, that this high level of Angiotensin 2 would have caused upregulation of his AT2 receptors.
The Renin Angiotensin System [RAS] has been previously thought of only as systemic endocrine system that is regulated at the renal level. Renal renin converts its circulating substrate angiotensinogen into Angiotensin 1 which is then converted to Angiotensin 2 by angiotensin converting enzyme. The Angiotensin 2 acts either through the AT1 or AT2 receptor in different tissues in the body including vascular smooth, vascular endothelium, and skin. [24,25].
It has now been shown that production of Angiotensin 2 also occurs at the tissue level with cell to cell signaling and no hormone secretion into the systemic circulation [26]. This is called a paracrine system. These are independently functioning RAS’s, and to be recognized as such, must have all the precursors and end products present in the tissue. They have been shown to be present in skin, vascular smooth muscle, and vascular endothelial tissue [23-25]. In the presence of high levels of systemic Angiotensin 2, the substrate angiotensinogen is upregulated in the paracrine system and this leads to increased local production of Angiotensin 2 [24,26]. This results in the systemic endocrine signal being amplified by causing local production of Angiotensin 2 at the tissue level. The signal is thereby both greater in magnitude and duration. In the renal circulation, this process has been shown to elevate the tissue levels of Angiotensin 2to a level that is 1,000 times greater than the level in the circulation [27].In addition to the paracrine system, there has now been identified an intracrine system whereby Angiotensin 2 is produced within the cell and stays within the cell and has a direct effect on the mitochondria [23,24,26]. The primary intracellular Angiotensin 2 receptor is the AT2 and not the AT1. This intracellular AT2 receptor is upregulated in the presence of high systemic Angiotensin 2 and the AT1 is not. Nitric oxide is produced in the cell via the AT2 receptor activation and its effect can be blocked with an Angiotensin antagonist [25].
It has been speculated that this signal amplification process and intracellular production of Angiotensin 2 may lead to disease at the cellular level [26]. I think that Rosacea is one such disease. Going back to the case presented, what was likely occurring after the initial renal injury was that the elevated levels of systemic Angiotensin 2 were upregulating AT2 receptor numbers in the vasculature of the patient’s face as well as amplifying the signal at the tissue and cellular level .After 6 weeks, this resulted in enough vasodilation and inflammation to produce clinically apparent Rosacea.
Hypertension is the world’s most prevalent cardiovascular disorder, with approximately one third of adults affected in most worldwide communities [27]. Hypertension has been shown to be a frequent comorbid condition of Rosacea [28,29]. Not only do both disorders have their onset in young adulthood and continue as chronic conditions, but the severity of Rosacea is tied to the severity of the hypertension. That is, moderate to severe Rosacea has a greater association with hypertension than does mild Rosacea [29]. This observation parallels what we saw in the case presented, whereby the Rosacea was most severe when the blood pressure was highest and moderated as the blood pressure came under better control.
No cause for this association has been proposed, but I believe there are two ways to explain this comorbid severity association. One is when there is moderate or high systemic renin/angiotensin activity and the other is when the systemic levels of renin and, therefore, Angiotensin are normal but there is an increased sensitivity of the target tissue to the Angiotensin.
In any hormone system, there is a range of what would be considered normal levels and some individuals will fall at the lower end and some at the higher end. Angiotensin Converting Enzyme (ACE) inhibitors are drugs that block the conversion of Angiotensin 1 to Angiotensin 2. These drugs can only be effective if the levels of renin/angiotensin are high enough to be reduced and, thereby, produce a reduction in blood pressure. People with relatively low levels of systemic renin activity will not respond to them. It is well recognized that the white population tends to respond well to these drugs and the black population less so. This observation is reflected in the treatment guidelines for hypertension that are issued by all the major advisory groups that advise ACE inhibitors in whites and other drugs in blacks. Steinhoff has identified fair skinned people of European descent as being more likely to be affected by Rosacea than African Americans [30]. He further said that African Americans are more likely to be affected if one parent is of northern European origin.
One can see that the response to ACE inhibitors as a treatment for hypertension parallels the likelihood of developing Rosacea. The responsive white population tends to develop Rosacea and the unresponsive black population does not. If one responds to an ACE inhibitor, it is reasonable to conclude that the systemic level of renin/ angiotensin in that individual is at least moderately elevated. Years of exposure to these elevated levels of systemic Angiotensin hormone will first lead to upregulation of the number of AT2 receptors in the vasculature of the skin. Signal amplification in the paracrine and autocrine systems will follow, and the net result will be a sustained level in the skin of bradykinin, nitric oxide, and the cascade of inflammatory mediators that are outlined in Figure 1. Ultimately, the phenotypic expression of this will be Rosacea in susceptible individuals.
The second way to develop Rosacea in association with hypertension is when the renin/angiotensin levels are in the normal or low normal range, but there is increased sensitivity of the target tissue to the Angiotensin. Williams expressed in 1994 that the majority of patients with essential hypertension have an endocrine basis for their elevated blood pressure but that the problem is not one of overproduction of circulating hormones; it is rather a change in the response of the target tissue to the hormone [31]. Carey, in 2015, stated that one of the genetic factors being studied in hypertension is the “inappropriately high activity of the RAS” at the paracrine level [27]. He used an example of this in mice that had an over expression of angiotensinogen (the precursor substrate for Angiotensin) in the paracrine RAS in their kidneys. When these mice were bred with transgenic mice that expressed human renin systemically, they developed “a major increase in blood pressure”. This occurred even though the circulating levels of Angiotensin 2 were normal. Carey also stated that he felt this finding in rats would also likely apply to humans. Genetic variability in Rosacea is a well-established concept and has been used to explain the phenotypic variation of the disease.[30,43] Genetic variability is what determines the extent and responsiveness of one’s paracrine and autocrine systems in the facial vasculature.
When one accepts this concept of genetic variability, it becomes easy to envision that there are people who will develop Rosacea in the presence of normal renin levels, in the same way that they develop hypertension under these circumstances. Both are related to a relative over activity of the paracrine/autocrine RAS. It explains why these disorders are linked together in a comorbid severity manner.
Sunlight, sweating, stress (adrenaline), heat, exercise, and alcohol have all been shown to exacerbate Rosacea. This relationship is not explainable via the AT2 receptor and another pathway must exist. The kallikrein enzyme is an enzyme found in skin which converts kininogen to bradykinin [17]. The area of greatest concentration of these enzymes is in the skin of the face as opposed to other regions in the body. The structure within the skin that harbors the highest concentrations of kallikrein enzyme is the sweat gland, and high activities of the enzyme have been demonstrated in various studies under the conditions aforementioned. Bradykinin is the product produced in this reaction, and as we have seen previously, this results in the generation of nitric oxide and a cascade of inflammatory mediators [17,32-37]. This is outlined in Figure 1.
In 2004, azelaic acid was FDA approved for the treatment of type 2 Rosacea but not type 1. Its mechanism of action was unknown at that time. In 2012 azelaic acid was proven to be a kallikrein inhibitor [38-41]. This validates the kallikrein arm of the Rosacea pathway that I proposed in 2000 [42] and leaves only the Angiotensin arm to be proven. That one arm is valid and the other is invalid seems unlikely, as neither one can stand alone as an explanation of the disease, but together, they do.
I view the kallikrein and Angiotensin pathways as being independent of one another, but their relationship may, in fact, be more than just that of a shared common pathway. We know that kallikrein inhibition alone does not treat type 1 Rosacea, and none of the currently available drugs on the market today has been approved to treat the persistent facial erythema that plagues Rosacea sufferers [2]. Antagonism of the AT2 receptor may well be effective in treating the features of type 1 based on what I have presented in this paper. But could it also treat the PFE that can develop after years of type 2 Rosacea? I think the answer is yes.
Many patients who start out as type 2 initially, will evolve into type 1 clinically and no longer respond to type 2 management [4,43]. It has been proposed that this is due to either extensive disease burden or the coalescence of individual papules and pustules, or both. This results in a background erythema that does not resolve with treatment of kallikrein activation. The obvious question, then, is why does the erythema not resolve if kallikrein activation alone is responsible for this phenotype? The answer may be that after prolonged kallikrein activation in the skin, Angiotensin activation takes over and PFE results.
We have seen that injury to vascular endothelium upregulates the number of AT2 receptors in skin. It is conceivable that kallikrein activation in the skin of susceptible individuals could not only initiate Type 2 Rosacea but may, in fact, eventually act as a form of “dermatologic trauma”. We have seen in studies that trauma resulting in upregulation of AT2 receptors in the skin was induced by scalpel in two studies [20,21] and balloon angioplasty in another study [19]. We know that trauma to the human body can be delivered in many forms and is certainly not limited to the ones used in these studies. Therefore, I would suggest that the reason for the persistence of facial erythema in treated type 2 Rosacea patients is because of upregulation of the AT2 receptor due to the repetitive kallikrein trauma, which then leads to paracrine and autocrine amplification of the Angiotensin system. This would explain how Type 2 patients become unresponsive to a kallikrein inhibitor and how the persistent facial erythema is caused. It also leads to the possibility that an AT2 receptor antagonist could resolve this situation.
Rosacea is a disease that needs effective treatment. Millions of people suffer from it and the burden is substantial with loss of productivity at work, loss of self-esteem resulting in difficulty with relationships, and loss of enjoyment of life [44-46].
Current therapies include topical and oral antimicrobials, topical vasoconstrictors, topical and oral retinoids, and phototherapy. Although each has proven to have benefit in selected patients, none has led the way as a highly effective therapy. This is likely because the key mediators of the disease are not being targeted.
Topical azelaic acid is a Rosacea therapy which is distinct from those listed above. It is a kallikrein inhibitor and identifies the enzyme as a key mediator of this disease. This provides solid support that one arm of the pathway proposed in this paper is correct, and I think increases the likelihood that the second arm, the Angiotensin arm, is also correct.
The RAS and the AT2 receptor specifically have never been implicated in Rosacea. This theory has evolved because a patient model presented itself and has served as a compass to guide my thought in looking at this disease. This situation is a rare opportunity in medicine and the information that it has provided should not be overlooked.
Rosacea is, I believe, a disease driven by Angiotensin activation and exacerbated by kallikrein activation. These are 2 independent arms that share a common pathway through bradykinin activation but relate to one another through crosstalk. In 2007, Dr. Ronald Marks stated that Rosacea may in fact be a collection of several diseases, all manifesting the same set of physical signs and acting through a common pathway. He expressed that the goal of research was to find a central unifying theme that can explain the pathophysiology of Rosacea [47]. I believe this pathway satisfies that goal, and in doing so, may lead to a cure of this disease by developing a novel medication with a specific target.
Download Provisional PDF Here
Aritcle Type: CASE REPORT
Citation: Bolla J (2020) Rationale for the Treatment of Rosacea with an Angiotensin At2 Antagonist. J Clin Cosmet Dermatol 4(3): dx.doi. org/10.16966/2576-2826.154
Copyright: © 2020 Bolla J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access