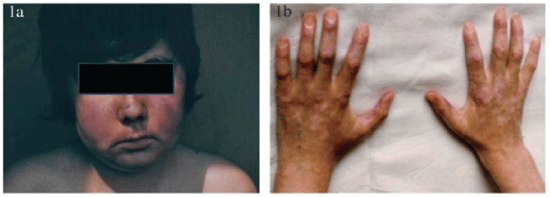
Figure 1: JSLE clinical manifestation (a -skin, h -hands) in a boy

D Mozolova1 J Rovensky2*
1Pediatric Department of the Medical Faculty, Comenius University, Bratislava, Slovakia*Corresponding author: J Rovensky, National Institute of Rheumatic Diseases, Piestany institute of Balneology, Physiotherapy and Therapeutic Rehabilitation, University of St. Cyril and Methodius, Trnava, Slovakia, E-mail: rovensky.jozef@gmail.com
Juvenile systemic lupus erythematosus (JSLE) is an autoimmune multi systemic disease associated with vasculitis, connective tissue involvement and generation of auto antibodies directed against self-antigens. These auto antibodies, the production of which varies and depends on the activity of the disease, affect various systems and the extent and severity of the involvement arc decisive for the life expectancy of SLE patients [1].
Currently, also less severe SLE forms have been differentiated and treatment (although not causal) has been improved thanks to the progress of research at the molecular level, as a result of which the patients’ survivorship is longer than in the past [2].
It has been proved that SLE incidence rate is higher in women (9:1 female-to-male ratio) and this finding is almost the same in JSLE [3]. The disease most commonly manifests itself from 2nd decade, primarily during puberty (Figure 1). Its onset in 1st decade is rare (Figure 2). Later in life (in old age) the SLE incidence rate generally decreases probably due to decreased activity of immune mechanisms.
Figure 1: JSLE clinical manifestation (a -skin, h -hands) in a boy
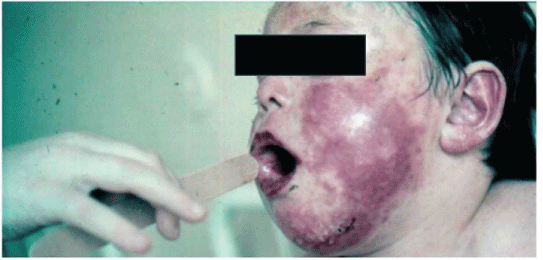
Figure 2: JSLE clinical manifestation in a boy
There are multiple genetic (HLA antigen system), hormonal (estrogens in females), and environmental (exposure to sunshine, smoking, certain medications) factors that are known to influence the development and nature of the disease.
The contributing factors for JSLE include in addition to pathological immune mechanisms (T and B cell systems), also other factors, as mentioned above. The genetic predisposition was suggested also in our group of patients, where SLE was diagnosed in identical twins and one of them had positive SLE immunological findings although clinical symptoms were not clear, yet.
A wide range of symptoms in JSLE correlates with involvement of multiple organs and includes skin symptoms, disorders of kidneys, heart, lungs, liver, CNS as well as of the hematopoietic system. The symptoms are of various intensity, they occur at different time intervals and their recurrence is sometimes unpredictable. The changing symptomatology and the latest findings also from other medical disciplines have led to revision of major and minor diagnostic criteria allowing a more accurate diagnosis of this disease [4].
Laboratory tests show high FW (ESR) values, sometimes bi- or pancytopenia, autoimmune hemolytic anemia, including abnormal urine and liver tests. Immunological parameters often include positive auto antibodies-ANA, anti-DNP, dsDNA, but also anti-Ro and anti-La, Sm/ RNP from the group of ENA antibodies. Specificity and sensitivity of individual auto antibodies often vary and the tests must be repeated to detect dynamics of the disease at particular time intervals.
SLE may be in general associated with the secondary Sjogren’s syndrome, which is exceptional in children, but also with the secondary antiphospholipid syndrome that may have catastrophic consequences. However, it is also rare in children and we have not encountered it in the group of patients (including girls) [5].
Clinical features of the disease gradually change from unspecific symptoms (fatigue, fever, arthralgia, myalgia, hair loss, exanthema and others) to a fully developed clinical manifestation, including severe symptoms of involvement of individual organs-glomerulonephritis or nephritis with nephrotic syndrome, Exsudative pericarditis, myocarditis, Exsudative pleuritis, symptoms of polyserositis, as well as symptoms of CNS involvement [6]. Convulsive seizures, psychosis, disorientation-that may have fatal consequences. An unnoticed and untreated disease has a rapid progress, while if diagnosed and adequately treated it has a more favorable prognosis [6].
Our group of patients with JSLE covering the period of 1970-2014, comprised 30 children with JSLE in the 5-18-year age group, of these 7 boys and 23 girl s. The characteristics of involvement of individual organs in boys are shown in Table 1. Lupus-like syndrome with deficiency of the Clq complement component was found in 2 boys and I girl from two Romany consanguineous couples (Figures 3-5) of the group of 23 girls, three patients died (once severe viral infection, once Libman-Sacks endocarditis and once multiorgan failure following relapse of the disease) [4,7,8]. As individual patients were admitted to hospital at different times when certain tests were not available yet, the data on some of them are incomplete. At the time when we could admit to hospital and follow up only children up to the age of 15 we had no feedback or later details about the progress of the patients’ disease or death. Table I shows patients who died.
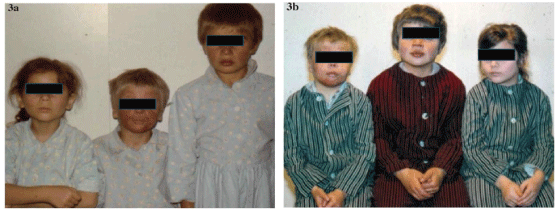
Figure 3: Patients with lupus-like syndrome and deficiency of Clq complement component from two consanguineous couples at admission (a) and follow-up (b) [12].
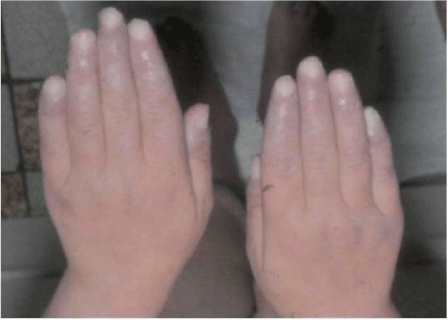
Figure 4: Secondary Raynaud”s phenomenon and vascu litis in a patient with lupus-like syndrome and deficiency of Clq complement component.
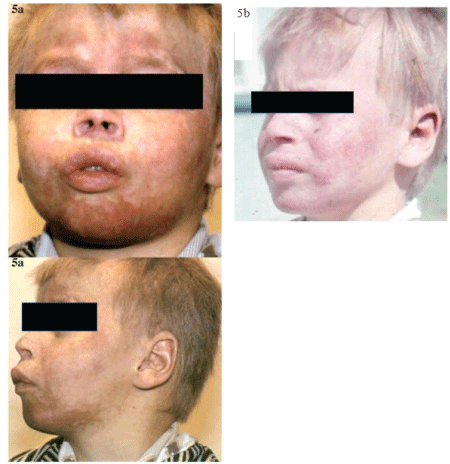
Figure 5: Lupus like syndrome in a boy [12].
(a) exanthema, alopecia, ulcerations at the nasal opening and auricles in a boy with a deficiency of Clq component; with a simultaneous Exsudative pericarditis, hepatopathy, nephropathy, high humoral activity, leukopenia, positive ANA and LE phenomenon, (b) partial improvement of exanthema after corticosteroid treatment.
| Patient | Age at diagnosis | Kidney | Heart | lungs | CNS | Hematology | Skin | Age at death |
| 1 | 10 | GLN | Le penia (Leukopenia) | Desrete LE erythema | 15 | |||
| 2 | 8 | Exsudative pericarditis | Exsudative pleuritis | Convulsions | Le penia | 12 (Stevens- Johnson syndrome) | ||
| 3 | 5 | GLN | myocarditis | Exsudative pleuritis | pancytopenia | LE erythema (Lupus erythematosus) |
8 | |
| 4 | 14 | Nephritis with nephrotic syndrome | ||||||
| 5* | 6 | GLN | Exsudative Pericarditis myocarditis | Exsudative pleuritis | Le penia | Generalized erytthema | 8 | |
| 6* | 8 | Exsudative Pericarditis | Le penia | Raynaud, LE erythema | ? | |||
| 7 | 15 | GLN | Exsudative Pericarditis | Exsudative pleuritis | Convulsions | pancytopenia | LE erytthema Vasculopathy | 15 |
Table 1: Overview of involvement of individual organs in boys with SLE
Patients received full corticosteroid dosage that was gradually reduced at particular time intervals based on improvement of clinical features (Figure 5) and before and after treatment decrease of laboratory parameters Regression or immunological Findings were relatively lower as compared to other laboratory indicators. Other symptomatic and replacement therapies (Ca, K, vitamin D, gastro protective drugs or antibiotics to treat infections) were regularly provided to all children during their hospital stay (although adherence to the therapy mainly by Roma children alter their discharge from the hospital was problematic). Antimalarials were administered to children based on the instantaneous activity of the disease. Biological treatment of SLE could not be applied in our group, other treatment rather than cortiosteroids e.g. mycophenolate mofetil, methotrexate for arthritis as the group of boys was followed long before it was reported [9]
The above mentioned facts show that JSLE is a severe, life-threatening disease that may occur also in boys, and should be taken into account in differential diagnosis. Our patients fulfilled criteria for SLE they are fever, cachexia syndrome arthritis, myocarditis, Pneumonitis glomerulonephritis, damage of skin, ANA positivity, Leukopenia, hyperimunoglobulinemia during course of disease [4,10,11]. In our group with JSLE from 1970-2014 we have 30 Childs with JSE in age 5-18 years old/23 girls and 7 boys. Deficiency C1q we found in 1 girl and 2 boys from consanquinal matrimony gypsy etnicum. Patients fulfill criteria for SLE in adults, and we found deficiency of C1q [13].
Download Provisional PDF Here
Article Type: Short Communication
Citation: Mozolova D, Rovensky J (2017) Juvenile Systemic Lupus Erythematosus in Boys. J Clin Case Stu 2(6): doi http://dx.doi.org/10.16966/2471-4925.159
Copyright: © 2017 Mozolova D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access