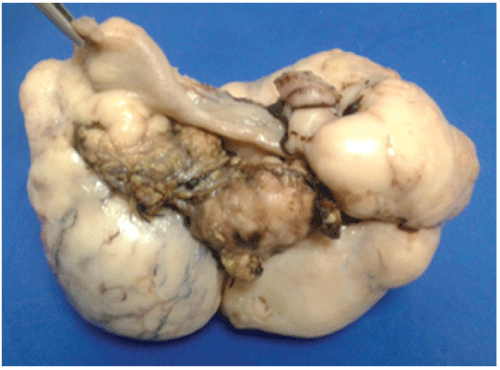
Figure 1: Right ovary tumor 20 × 12 × 10 cm

Guillermo Padrón Arredondo1* Adianez Oramas Rojo2 Renán Baqueiro Canto 3
1General Surgeon and Endoscopist*Corresponding author: Guillermo Padrón Arredondo, MD, Cerrada Corales 138, Residencial Playa del Sol. Solidaridad, Playa del Carmen, Quintana Roo, México, Tel: 01-984-876-2267; E-mail: gpadronarredondo@hotmail.com
Introduction: Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH) affects about 4,500 women around the world. This syndrome is a malformation of the female genitalia due to an embryonic development interrupt of the Müllerian (paramesonephric ducts) between the 5th and 6th week of embryonic development. On the other hand, the patients are chromosomally, phenotypically and endocrinologically normal.
Clinical Case: 10-year-old female who started with 4-days abdominal pain which on physical examination had normal phenotype, His laboratories were: Liver Function Tests: lactic dehydrogenase (LDH) 1876 U/l (103-227); Alkaline Phosphatase ALP 395 U/l (64-306 U/l). EGO: leukocytosis 15-20 (<10 × c) Abundant bacteria, erythrocytes 30-40. Embryonic carcinoembryonic antigen (-), normal Human Chorionic Gonadotropin. Ascites liquid: pH 9, DHL 2225 U/l, Hb (+++), 100-150 leukocytes × C, countless erythrocytes. An exploratory laparotomy a bilateral ovarian tumor with uterine agenesis (Müllerian alteration) was found, which were resected to obtain a tumor of 550 g of 20 × 12 × 10 cm. Computed tomography is then performed. The histopathology service reports dysgerminoma. The patient satisfactorily recovers from her surgical intervention and is sent to the oncology department for her tumor treatment.
Discussion: In our case postoperative computed tomography showed no renal damage and the imaging study (CT) was performed after its surgical intervention; genetic origin has been extensively documented in multiple studies. Our case presented cancer developed on the right ovary and incipient cancer on the left ovary as well as retroperitoneal metastatic lesion below the right kidney. This case is unfavorable because of its association with ovarian cancer at an early age and metastatic to retroperitoneum.
Mayer-Rokitansky-Kuster-Hauser syndrome; Vaginal agenesis; Müllerian agenesis; Ovarian dysgerminoma
Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH) affects about 4,500 women around the world, this syndrome is a malformation of the female genitalia due to an embryonic development interrupt of the Müllerian (paramesonephric ducts) between the 5th and 6th week of embryonic development. On the other hand, the patients are chromosomally, phenotypically and endocrinologically normal. This syndrome is subdivided into type A with symmetrical uterine remnants and with normal fallopian tubes, and type B (atypical) with asymmetric uterine outbreaks, abnormal fallopian tubes and various abnormalities of other organs and systems [1]. Type 1 MRKH syndrome is less frequent and type 2 anomalies include: renal (unilateral agenesia, ectopia of kidneys or horseshoe kidneys), Skeletal (particularly vertebral as in Klippel-Feil syndrome, fused vertebrae, scoliosis), Hearing defects and rarely cardiac anomalies and limb anomalies such as syndactyly or polydactyly) [2].
Anomalies associated with this syndrome. Anomalies of the urinary tract are observed in 15% of cases. The most frequent are:
Due to the frequency of anomalies in the urinary system, it is common for the patient to require ultrasound, intravenous and pyelotac studies to rule out associated malformations.
Skeletal anomalies are found in 12 to 50% of patients. Most are vertebral alterations:
Sometimes accompanied by alterations on the limbs, eg: clinodactyly, polydactyly, and hypoplasia of the radius, scaphoid or trapezium. Other anomalies are reported, such as the association of Müllerian agenesis with malformative syndromes such as Klippel-Feil (congenital fusion of two or more cervical vertebrae that causes decreased movement and shortening of the neck) or MURCS syndrome (aplasia mülleriana, aplasia renal and cervicothoracic dysplasia caused by alterations in the corresponding somites) [3].
This MRKH syndrome has long been considered an occasional anomaly, but the literature and family cases support the etiology of the existence of a specific genetic substrate and at present this syndrome seems to be transmitted as an autosomal dominant character with incomplete penetrance of variable expression [4].
10-year-old female who started with 4-day abdominal pain which on physical examination had normal phenotype, height 1.37 m, weight 26 kg with moderate dehydration, scales on skin, scalp and ears and hypopigmented leather spots scalp, generalized erythema and conjunctivitis. His laboratories of admission were: Normal blood chemistry: normal Hematical biometry; Liver Function Tests: lactic dehydrogenase (LDH) 1876 U/l (103-227); Alkaline Phosphatase ALP 395 U/l (64-306 U/l). EGO: leukocytosis 15-20 (<10 × c) Abundant bacteria, erythrocytes 30-40. Embryonic carcinoembryonic antigen (-), normal Human Chorionic Gonadotropin. Ascites liquid: pH 9, DHL 2225 U/l, (Hb (+++), 100-150 leukocytes × C, countless erythrocytes. An exploratory laparotomy is performed with a diagnosis of probable complicated appendicitis. A bilateral ovarian tumor with uterine agenesis (Müllerian alteration) was found, which were resected to obtain a tumor of 550 g of 20 × 12 × 10 cm, figures 1 and 2. Computed tomography is then performed. (Figures 3 and 4) Surgical specimen is sent to the histopathology service that reports dysgerminoma. (Figure 5) The patient satisfactorily recovers from her surgical intervention and is sent to the oncology department for her tumor treatment.
Figure 1: Right ovary tumor 20 × 12 × 10 cm
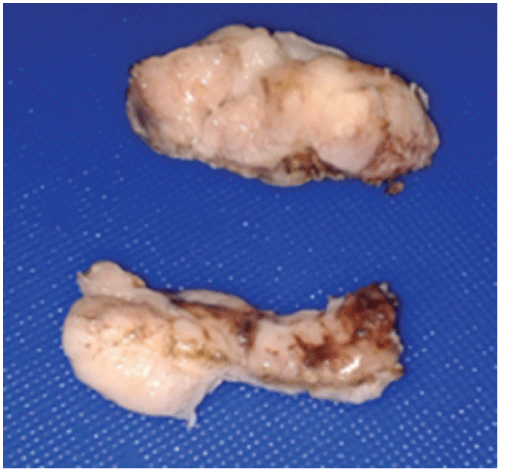
Figure 2: Left ovary tumors
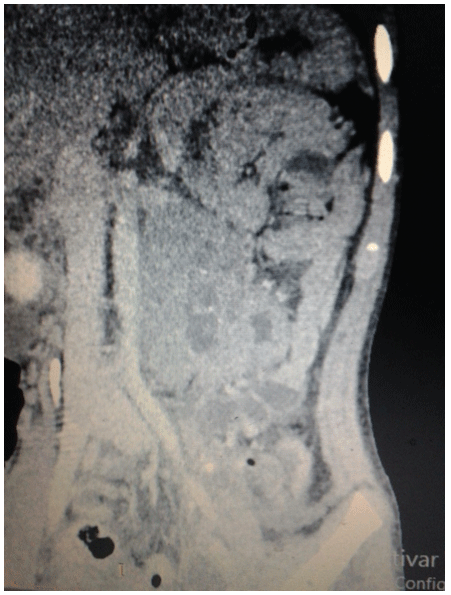
Figure 3: Contrast tomography after the surgical event in a coronal section where it is observed: at the retroperitoneum level and below the left kidney: an irregular-shaped, oval-shaped, heterogeneous image with soft tissue density and hypodense center (by necrotic tissue) and scarce calcifications, with heterogeneous enhancement of the contrast medium with measures of 75 × 56mm and displacing the left kidney towards superior, without loss of the fat interface of both.

Figure 4: Contrast tomography in sagital cortex observing: in uterine topography there is absence of the uterus, as well as proximal portion of a vaginal cavity. Urinary bladder (anterior) and lower portion of the rectum (posterior and posterior sacral vertebras are observed).
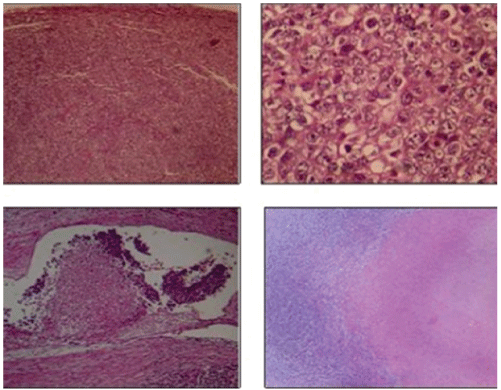
Figure 5: Malignant neoplasm of germinal strain, with solid pattern, composed of septated lobes (connective tissue septa). The neoplastic cells are large, from eosinophilic cytoplasms to amphiphiles, the nuclei have irregular contours and evident nucleoli. Are accompanied by abundant mitotic figures, inflammatory infiltrate, cellular detritus and necrosis in 30% of the tumor. In addition, there is lymphatic-vascular invasion.
Kamal NM et al. [5] reported a case similar to ours where they located an ovary dysgerminoma in a 7-year-old girl with transvaginal bleeding but without association with MRKH syndrome. Similarly, Miura R. et al. [6] reports a case of dysgerminoma in an ectopic ovary associated with WAGR syndrome (Wilms’ tumor, aniridia, genitourinary anomalies and mental retardation) of genetic origin without association with the MRKH syndrome.
It is known that malignant ovarian tumors such as disgeminoma may be associated with patients with pure gondadal dysgenesis (46 XY B/L streak gonads), mixed gonadal dysgenesis (45 X / 46 XY, U / L streak gonad, C / L testis ) and testicular feminization syndrome [7].
In a study of Oppelt PG et al. [8] in 284 patients with MRKH syndrome and malformations found complete vaginal and cervical atresia in all of them with variable malformations in uterus and appendages; in addition, they observed a variety of malformations, mainly renal. In our case postoperative computed tomography showed no renal damage. BjørsumMeyer T et al. [9] reports an interesting case with multiple malformations: vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, esophageal atresia, renal defect, limb defects in association with MRKH syndrome. Similarly, our case did not present associated malformations of any type, which leads to MRKH type 1.Only Mishina A et al. [10] report a case similar to ours because is very rare.
On the other hand, Fiaschetti V et al. [11] emphasize that Nuclear Magnetic Resonance (NMR) is the non-invasive choice study with a higher sensitivity and specificity than ultrasound and is a complementary tool of diagnostic laparotomy when this diagnosis is suspected previously which was not suspected like our case until a diagnostic laparotomy was performed. On the other hand, Yoo R-E et al. [12] in their analysis on the usefulness of NMR vs. diagnostic laparoscopy found that the former is superior because it can detail the underlying abnormalities much better than laparoscopy. But, Dragusin et al. [13] in their study establish that laparoscopy is superior to MRI by defining more accurately the anatomical characteristics of MRKH syndrome.
Acién P et al. [14] consider that for the best diagnosis and treatment of these abnormalities the use of an embryological system to catalog these abnormalities correlates with clinical presentation, in addition to the use of NMR. Drummond JB et al. [15] conducted a study on β-catenin in the DNA of patients with MRKH in order to locate some genetic defect without finding evidence of such defect.
Genetic origin has been extensively documented in multiple studies and cases, for example: Bernardini et al. [16] reports two cases with identical deletion of chromosome 17q12. Nodale et al. [17] found that a consistent over expression of the HOXC8 gene could be a compensatory mechanism to decrease the expression of other genes, which may limit the occurrence of other malformations, especially those involving the renal system and then produce a phenotype (type 1MRKH).
Morcel et al. [18] studying DiGiorge disease (DGS) found deletions in four probands within the four chromosomal loci 4q34-qter, 8p23.1, 10p14 and 22q11.2 implicated in almost all cases of DGS syndrome, concluding that uterovaginal aplasia seems to be an additional feature of the broad phenotypic spectrum of the DGS.
Ma W et al. [19] in their study successfully identified two single nucleotide polymorphisms (SNPs) of susceptibility (WNT9B and PBX1) associated with the risk of MRKH syndrome, both separately and interactively. The discovery of an epistatic effect of four genes (AMH, PBX1, WNT7A and WNT9B) on MRKH syndrome provides new information for the elucidation of the genetic mechanism underlying the etiology of MRKH syndrome.
The incidence of MRKH/MURCS syndrome association has been undervalued and until recently has been seen as a specific and sporadic female disorder. But the isolated features of the triad of major malformations, including renal agenesis and / or skeletal defects, were not investigated in all relatives of the probands, including men who may also be affected. This is understandable given the incomplete degree of penetration, variable expressiveness and similarities of this syndrome with other genetic disorders [20].
As for the complications associated with MRKH, these are diverse among them we can find: leiomyoma in a rudimentary uterus. Rawat et al. [21]Nath et al. [22] countered an association with psychotic conditions in these patients. On the other hand, the consequences of having MRKH syndrome is related to a higher level of neuroticism compared to the general population. An important factor related to variables is the time elapsed from diagnosis to treatment. Women with MRKH syndrome should be supported with specialized psychiatric or psychiatric help from the time they are diagnosed [23].
Acién P et al. [24] report renal dysplasia and pulmonary hypoplasia. Sharifiaghdas F et al. [25] reports a case of transurethral coitus with urinary incontinence. Uçar MG et al. [26] likewise report urethral coitus and question whether vaginoplasty is sufficient to prevent urinary tract infections. Bae HS et al. [27] report the first known case of cancer in a supernumerary ovary in a patient with MRKH syndrome. Our case presented cancer developed in right ovary and incipient cancer in the left ovary as well as retroperitoneal metastatic lesion below the right kidney.
Rall K et al. [28] found a low frequency of acne in patients with MRKH despite high levels of hyperandrogenemia. With respect to the treatment of this syndrome, this will depend on the anomalies found: Saleem M et al. [29] performed a colpovaginoplasty to join the uterus with the introitus; surgery minimally invasive reconstructive plastic surgery (Neovagina Vecchietti) is a successful technique in MRKH syndrome [30,31].
Wagner A et al. [32] identified health care requirements and measures for the following areas: diagnosis during adolescence and organization of care, diagnostic reactions, functional infertility, psychological stress, and the threat of self-image. Contact with others and treatment of MRKH coping strategies are objectives to be taken into account during the transition from childhood to adolescence and adulthood in these patients.
Although there is still much to learn about the etiology of MRKH, there has been steady progress in recent decades with regard to efficient diagnostic modalities and proper medical management. Non-surgical approaches to the creation of aneovagine are at the heart of therapeutic options, and should also be recommended for most patients who undergo surgical treatment in order to preserve functional outcomes.
Treatment in childhood and early adolescence is not recommended due to the rate of unacceptable complications and because a full understanding and commitment of the patient is required to obtain optimal results. Fertility options through IVF using autologous oocytes and a surrogate gestational carrier are increasingly available at reference centers. Continuous monitoring of the psychological well-being of these patients should be considered [33]. Our case is unfavorable because of its association with ovarian cancer at an early age and metastatic to retroperitoneum.
Download Provisional PDF Here
Article Type: Case Study
Citation: Padrón-Arredondo G, Rojo Oramas A, Canto Baqueiro B (2017) Ovary Dysgerminoma in Girl of 10 Years-Old Associate with MayerRokitansky-Kuster-Hauser Syndrome (Mrkh) Type 1. A Unusual Condition. Clinical Case. J Clin Case Stu 2(2): doi http://dx.doi.org/10.16966/2471-4925.142
Copyright: © Padrón-Arredondo G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access