Case Report
A 35 year old man presented in childhood with global developmental
delay, hypotonia and a Marfanoid phenotype with dolichocephaly, long
narrow face with malar hypoplasia and retrognathia, high-arched palate
and dental crowding, chest asymmetry, severe scoliosis and pes planus
(Figure 1A). There were no ocular findings of Marfan syndrome (MS) and
no known relevant family history. Investigations revealed normal plasma
amino acids and urine organic acids, normal total plasma homocysteine
and normal fragile X syndrome results.
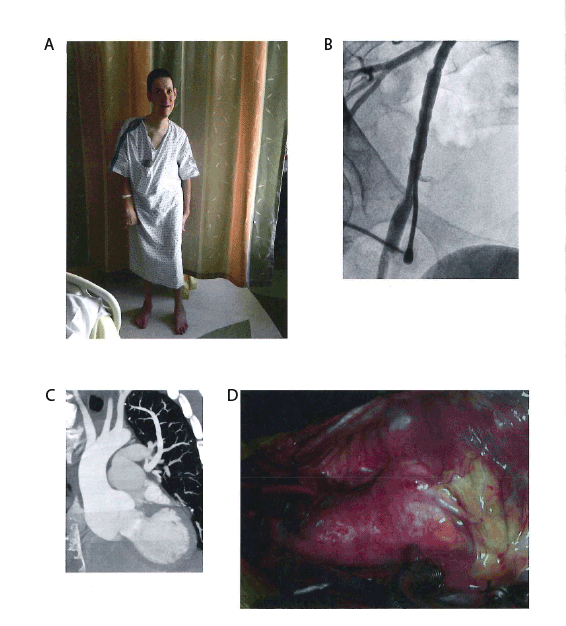
Figure 1: Phenotypic Features (A) With permission, photograph of patient demonstrating Marfanoid habitus. (B) Fluoroscopy image of the femoral
artery demonstrating a “beaded” appearance. (C) Computed tomography image (coronal plane) demonstrating dilated aortic root and pulmonary
artery. (D) Intraoperative photograph demonstrating syndromic aneurysmal aortic root. Cranial is to the left and caudal to the right.
At this visit, there was progressive shortness of breath, moderate
aortic regurgitation and progressive dilatation of aortic root dimensions
(3cm at 8 years, 4.6 cm by 20 years and 5.6 cm at 35 years on serial
transthoracic echocardiography confirmed by computed tomography.
The latter also showed dilated pulmonary artery sinuses to 4.1cm (Figure
1B). During his work up for aortic root surgery, a coronary angiogram via
transfemoral access revealed a “string of beads” appearance of the femoral
artery (Figure 1C) typical of the medial fibroplasia type of fibromuscular
dysplasia [1].
Cytogenetic microarray (Combimatrix, Irvine CA) analysis
revealed a de novo unbalanced karyotype 46, XY, der(4)t(4;10)
(q34.3;p12.1)dn.arr [gh19] 4q34.3q35.2(179,589,516-191,154,276)
x1,10p15.3p12.1(0-29,304,999)x3.
The derivative chromosome 4 resulted from an unbalanced translocation
between the long arm of chromosome 4 and the short arm of chromosome
10, at bands 4q34.3 and 10p12.1, respectively. The resulting net genetic
imbalance of the der(4) is monosomy for the distal region of chromosome
4from 4q34.3 ≥ q35 and trisomy for the region of chromosome 10from
10p12.1≥ p15 (Figure 2). These findings were confirmed by fluorescent
in situ hybridization (FISH) analysis (data not shown). Both parents had
normal microarray and FISH analyses.
Therefore, our provisional diagnosis was an adult male with severe
intellectual disability, aortic root dilatation and other vascular anomalies,
and a Marfanoid habitus resulting from an unbalanced karyotype. DNA
sequencing of the ACTA2, FBN1, TGFBR 1 and 2, TGFB2, COL3A1 and
SMAD3 genes in a core vascular aneurysm panel revealed no known
disease-causing mutations (Collagen Diagnostic Laboratory, University of
Washington and Seattle).
The patient successfully underwent a tricuspid aortic valve sparing
procedure (David’s reimplantation). Intraoperatively, his aortic root and
pulmonary artery dilatations appeared very similar to the dilatation seen
in MS or Loeys-Dietz syndrome (Figure 1D). Histologically cystic medial
necrosis was observed in the resected specimen. Some features of both
these disorders were lacking. The patient did not meet the 2010 Ghent
criteria for MS [4] and DNA studies of known candidate genes proved
negative.
Global developmental delay, hypotonia and craniofacial features seen
in our patient have been described in patients with 10p trisomy [2],
whereas distal 4q monosomy has been associated with milder phenotypes
[3]. We believe that this derivative chromosome is, to our knowledge,
unique. Detailed review of the many genes known to be mapped to the
unbalanced segments of 10p and 4q using OMIM and other databases
(Decipher, UCSC Genome Browser) revealed no candidate genes,
and no genetic disorders associated with a Marfanoid phenotype [4],
fibromuscular dysplasia, or syndromic aortic root dilatation. However
many genes mapped to the 10p and 4q regions of interest have no known
phenotype as yet. This would suggest either a new syndrome involving a
candidate gene mapped to 10p or 4q with a new function or, there may
be a gene mutation in a chromosome region elsewhere in the genome
associated with aortopathy and developmental disability as previously
described [5].
In conclusion, patients with chromosome aneuploidy associated with
syndromic features and /or Marfanoid phenotype should be monitored
for aortic root dilatation and should be surgically treated between 4.2 cm
(Loeys-Dietz) to 5cm (MS), depending upon rate of growth. Parents and
siblings of affected individuals should be screened with echocardiography
and microarray analysis.
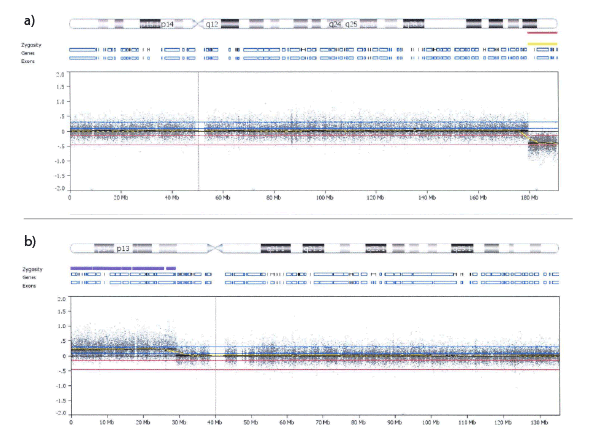
Figure 2: Cytogenetic microarray of a) Chromosome 4, showing the deletion of 4q34.3 ≥ qter, b) Chromosome 10, showing the duplication of 10p12.1 ≥ pter
Download Provisional PDF Here
Article Information
Article Type: Case Report
Citation: Shah P, Dawson A, Tam J, Semaniuk N,
Hovanes K, et al. (2016) Distal Trisomy 10p and 4q
Monosomy: Associated with Marfanoid Features,
Syndromic Aortic Root Dilatation and Intellectual
Disability. J Clin Case Stu 1(1): doi http://dx.doi.
org/10.16966/2471-4925.103
Copyright: © 2016 Shah P, et al. This is an
open-access article distributed under the terms
of the Creative Commons Attribution License,
which permits unrestricted use, distribution, and
reproduction in any medium, provided the original
author and source are credited.
Publication history:
Received date: 9 Dec 2015
Accepted date: 24
Dec 2015
Published date: 5 Jan 2016
